
Translate this page into:
Bioequivalence study of two formulations of rivaroxaban in healthy adult subjects under fasting conditions
*Corresponding author: Evelyn Pena, MSc MD Department of Clinical Research, Industrias Biocontrolled C.A. Laboratorios Leti S.A.V., Guarenas, Miranda, Venezuela. martinpena24@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Pena E, Inatti A, Martin XS. Bioequivalence study of two formulations of rivaroxaban in healthy adult subjects under fasting conditions. Am J Pharmacother Pharm Sci 2023;8.
Abstract
Objectives:
Oral anticoagulants exert their antithrombotic effect by disrupting the coagulation cascade. Rivaroxaban is the first oral agent to be developed that inhibits the coagulation process by binding directly to Factor Xa in a competitive manner. The aim of this study was to demonstrate the bioequivalence (BE) and safety of a generic formulation of rivaroxaban by comparing their pharmacokinetic (PK) parameters through statistical data and criteria of validation. Oral tablet formulations of 20 mg of a commercial product rivaroxaban reference (R) were tested against a generic product test (T) in 24 healthy adults under fasting condition.
Materials and Methods:
The study was an open label, balanced, randomized, two-treatment, two-period, two-sequence, single oral dose, and crossover study. Blood samples were collected pre-dose and at specified intervals up to 48-h post-dose to evaluate PK parameters by quantifying the concentration of rivaroxaban in plasma using a validated Liquid chromatography-mass spectrometry (LC-MS/MS) method of analysis. Statistics and confidence intervals (CIs) were calculated for BE purposes.
Results:
The geometric means of the T/R ratios and 90% confidence intervals (CIs) were: Cmax 87.80% (82.74 –93.12%), AUC0-t 85.96% (81.88–90.24%), and AUC0-∞ 86.13% (82.2–90.35%). All PK parameters are within BE acceptance range of 80–125% for demonstration of average bioequivalence.
Conclusions:
The study demonstrates the BE and well tolerance of both formulations of rivaroxaban in healthy subjects under fasting conditions.
Keywords
Bioequivalence
rivaroxaban
pharmacokinetics
anticoagulants
safety

INTRODUCTION
According to the World Health Organization, cardiovascular diseases are the leading cause of death globally, with an estimated 18 million deaths in 2020.[1,2] Thrombosis is the most common underlying pathology of the three major cardiovascular disorders: Ischemic heart disease, stroke, and venous thromboembolism (VTE).[3,4]
Advances on the molecular bases and mechanism of formation of thrombus and the coagulation cascade for clot formation have made possible to develop specific targets within the cascade as an alternative to heparin and vitamin K antagonists as anticoagulant therapy which are associated with several drawbacks.[5] New antithrombotic agents known as direct oral anticoagulants (DOACs) are now prescribed extensively in medical practice.[6]
Rivaroxaban, the first DOAC, was approved for clinical use in 2008 for the prevention of VTE. Following oral intake it binds directly and reversibly to factor Xa of the coagulation cascade.[5] It is also used to prevent stroke and systemic embolism in adults with non-valvular atrial fibrillation, for the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), as well as for the prevention of recurrent DVT, PE, and atherothrombotic events with acute coronary syndrome.[6-8]
The bioavailability (BA) of rivaroxaban is high (80–100%)[7,8] and it is affected by food ingestion where the maximum inhibitor effect is 2–4h after intake.[9] The half-life of the drug in young adults is 5–9 h and 11–12 h in older subjects. Absorption depends on the dose and ingestion of food. A daily dose of 15–20 mg taken with food significantly increases the oral absorption of rivaroxaban. Administration of rivaroxaban under fasting conditions reduces BA by approximately 66%.[10]
The research, development, and launch of new molecular entities as drugs is extremely costly. Hence, to recover costs of new products, innovator drug products are allowed to be patented for some time. However, to ensure the availability of sufficient cost-effective drugs for treatment of diseases, other pharmaceutical companies are allowed to market the generic versions of innovator drug products after their patents expire. Safety and efficacy of drug products is paramount, thus generic drugs must pass abbreviated clinical trials in the form of comparative bioavailability or bioequivalence (BE) studies and a strict regulatory control.[11-13]
The purpose of the present study was to assess and compare the PK profiles and safety of 20 mg of Xarelto® (Bayer Pvt. Ltd) as a reference (R) vs Asarap® (Laboratorios Leti S.A.V, República Bolivariana de Venezuela) as a test (T) formulation of rivaroxaban in healthy adult subjects under fasting conditions required for BE purposes. This study was conducted in India, by CRO ICBio Clinical Research Pvt, Ltd.
MATERIALS AND METHODS
Ethical Approval
The study was conducted ethically in accordance with the principles of the ICMR guidelines (2017), New Drugs and Clinical Trials Rules 2019 India, and adhered to the ethical principles of the Declaration of Helsinki, the International Conference on Harmonisation Good Clinical Practice Guidelines.[14-17] The study protocol was approved by an Independent Ethical Committee (ECR/141/indt/KA/2013/ RR-19), and certified by CDSCO/DGHS to ICBio Clinical Research Pvt, Ltd., BA/BE/2020/053. Study number: ICBio/020/0522.
Study design
The study was an open label, randomized, two-treatment, two-period, two-sequence, single oral dose, crossover, and BE study in healthy adult subjects under fasting conditions.
Tablets of Xarelto® (Batch BXJKGV1, expiration date 03/2024, Bayer Pvt. Ltd) were used as the reference (R) drug and Asarap® (Batch 006, expiration date 10/2023, Laboratorios Leti S.A.V, República Bolivariana de Venezuela) as the test (T) drug. Both study drugs contained rivaroxaban as the active pharmaceutical ingredient.
According to the randomization schedule [Table 1], a single dose of the study drug (T or R) was administered in each period. Subjects who received T product in period I were administered R product in period II and vice versa. Pre-screening period was 21 days. The study lasted for 10 days (October 31, 2022 to November 10, 2022) including 7 days washout considering the terminal half-life for rivaroxaban is between 7 and 17 h.[7,8]
| Subject | Sequence | Period I | Period II |
|---|---|---|---|
| 01 | RT | R | T |
| 02 | RT | R | T |
| 03 | RT | R | T |
| 04 | RT | R | T |
| 05 | TR | T | R |
| 06 | RT | R | T |
| 07 | TR | T | R |
| 08 | TR | T | R |
| 09 | RT | R | T |
| 10 | TR | T | R |
| 11 | TR | T | R |
| 12 | RT | R | T |
| 13 | RT | R | T |
| 14 | TR | T | R |
| 15 | RT | R | T |
| 16 | RT | R | T |
| 17 | RT | R | T |
| 18 | TR | T | R |
| 19 | TR | T | R |
| 20 | TR | T | R |
| 21 | RT | R | T |
| 22 | TR | T | R |
| 23 | TR | T | R |
| 24 | TR | T | R |
R: Reference Xarelto®, T: Test Asarap®
Subjects
Although the study was open to males and females based on the previous studies where the rivaroxaban pharmacokinetic (PK) in male and female subjects were not significant,[7,8] only male subjects fulfilled all the following inclusion criteria: Aged between 18 and 45 years, with good health based on the results of a complete clinical history and valid for 6 months before the start of the study; normal laboratory values as determined by medical history and physical examination at the time of screening; normal vital signs (blood pressure, pulse rate, and axillary temperature) and physical examination; prothrombin time, and activated partial thromboplastin time within normal range; creatinine clearance value of more than 50 mL/min; negative tests for hepatic transaminases, hepatitis B and C, human immunodeficiency virus and venereal disease research laboratory; normal 12-lead electrocardiogram (EKG) values and no >6 months before the start of the study; normal chest radiography and negative result in urine drug tests. Subjects with a body mass index (BMI) within a range of 18– 30 kg/m2 were included as well as non-smokers subjects or smokers who had not smoked at least 10 h before the start of the study. All the subjects were informed about the potential risks and the benefits of their participation such as blood laboratory test, EKG as well as travel and food expenses. They all signed the informed consent.
The exclusion criteria included a history of hypersensitivity to the study medication or to any other medication belonging to the study group or cardiovascular, renal, hepatic, metabolic, gastrointestinal, neurological, endocrine, hematopoietic, psychiatric, or other organic abnormalities; under medication that interferes with the quantification and/or kinetics of the medication under study or potentially toxic medications within 30 days before the start of the study; exposure to agents known as inducers or inhibitors of liver enzyme systems; taken any medication within 7 days or 7 half-lives before the start of the study; hospitalized for any reason or who were seriously ill within the 90 days prior to the study; received a research medication within 30 days before the start of the study; donated or lost 450 mL or more of blood within 90 days before the start of the study; recent history of drug abuse, including alcohol; consumed products such as cola drinks containing caffeine, theobromine, or theophylline in the 48 h before the study; grapefruit juice consumption in the 72 h before the study.
Drug administration and blood collection
All the subjects were fasted for at least 10 h pre-dose and 4-h post-dose. The subjects received standardized meals at 04.00, 08.00, 12.00, and 24.00 h after dosing in each period. During housing, the meal menu was same in both the periods (2500 Kcal) and drinking water was provided ad libitum.
Following an overnight fast of at least 10 h, subjects were scheduled for dosing as per the randomization schedule in each period [Table 1].
The study was conducted with 24 subjects for period I and 23 subjects for period II as one subject did not turn out for the second period and was considered a dropout. Single oral dose of either the T or R were administered with 240 mL of water at ambient temperature in each period under yellow monochromatic light.
A total of 20 × 6 mL blood samples were collected via cannula from each subject during each period while 24:00 and 48:00 h samples were collected by direct punction. The venous blood samples were withdrawn at pre-dose (00.00 h) and 00.17, 00.50, 00.75, 01.00, 01.50, 02.00, 02.50, 03.00, 03.50, 04.00, 04.50, 05.00, 05.50, 06.00, 08.00, 10.00, 16.00, 24.00, and 48.00 h.
Analytical procedure
The blood samples were collected in pre-labeled K2 ethylenediaminetetraacetic acid (EDTA) vacutainers and were centrifuged at 4000 rpm for 10 min at 2–8°C. Plasma was separated, labeled, and stored at −70 ± 5°C before analysis.
Plasma samples, calibration curve standards of internal standard (IS) Rivaroxaban D4, (Vivian Life Sciences Private Limited, Mumbai, India.), and quality control (QC) samples were thawed and vortexed for preparation and analysis. Aliquots of 250 µL plasma were mixed with 250 µL of extraction buffer and vortexed. Solid phase extraction on hydrophilic-lipophilic balance (HLB) cartridges was performed for sample preparation. After conditioning (1 mL of methanol), equilibrating (1 mL water) and loading the sample, cartridges were washed (1 mL of water followed by 1 mL of washing solution) and was dried. Cartridges were eluted with 800 µL of methanol and the eluate diluted with 200 µL of methanol. Controls samples were spiked with IS of over the concentration range of 1205.240–1.286 ng/mL Analytes, IS and QC samples were transferred to pre-labeled vials arranged in the autosampler at 10 ± 3°C. Analysis of rivaroxaban used an LC-ESI-MS/MS instrument (Shimadzu LCMS-8040, Mumbai, India). A BDS Hypersil C18 4.6 × 50 mm id 5 µm HPLC column was used (Thermo Scientific, Mumbai, India). And the mass spectrometer was operated in positive electrospray ionization mode. Identifications were based on multiple reaction monitoring transitions; m/z 436.2—269.7 for rivaroxaban and m/z 440.6—241.2 for the IS. The inter batch calibration standard precision was in range 0.94–2.75% and accuracy 97.17–101.71%.
Statistical and PKs analyses
The PK parameters calculated were maximum peak concentration (Cmax), area-under-curve (AUC) from time 0 h to the last measurable concentration (AUC0-t), AUC from time 0 to infinity (AUC0-∞), time to reach Cmax (Tmax), and elimination half-life (T1/2). PK and statistical analyses were performed using SAS version 9.3.1 Inc., Cary, North Carolina. USA. The log-transformed PK parameters were analyzed using a general linear model (Proc GLM of SAS® Mumbai, India). The sample size calculation for the study was based on intra-subject coefficient of variation (CV%)[7] for rivaroxaban according to Tao et al.[18] The difference of Law of least squares were calculated for the logarithmic (ln)-transformed PK parameters and the T/R ratios.
With the expected % CV for Cmax and AUC not exceeding 20% and the ratio falling within 95–105% (i.e., a true treatment difference of 5%), the study required 20 evaluable subjects to show BE with a power of 90% at 5% level of significance. Additional subjects were included in the study for possible dropouts/withdrawals. Thus, a total of 24 healthy subjects were sufficient to demonstrate BE between the test and reference products.
The geometric mean ratios (GMRs) of these primary PK parameters (T/R) and the 90% confidence intervals (CIs) were calculated for the determination of BE. Analysis of variance was applied on the logarithm-transformed PK values. BE between the test and reference formulations of rivaroxaban was demonstrated if the 90% CIs fell within the acceptance range of 80–125% for ln-transformed PK parameters Cmax, AUC0-t, and AUC0-∞.[11]
Safety assessments
The safety of two formulations was evaluated through the assessment of adverse events monitoring throughout the study. Vital signs were measured at baseline screening, and at the end of the study. Twelve-lead EKG and clinical laboratory such as urine analysis, blood biochemistry, and hematology evaluation were carried out at screening and 48-h post-study.
RESULTS
Subject disposition and baseline characteristics
PK and statistical analysis were performed for 22 subjects as two subjects were excluded: Subject 12 did not participate period-II and subject 01 had plasma levels <5% of reference medicinal product geometric mean AUC and was considered an outlier.
Participants baseline characteristics were mean age 33.13 ± 6.1 years, mean weight 70.80 ± 8.08 kg, mean height 1.7 ± 0.1 meters, and mean BMI was 24.57 ± 2.7 kg/m2 [Table 2].
| Age (years) | |
|---|---|
| Mean ±SD | 33.13 ± 6.11 |
| Range | (25-43) |
| Age Group | N/% |
| 18-40 | 19/86.36 |
| 41-64 | 3/13.64 |
| Total | 22/100% |
| Sex M/F | Male/100% |
| BMI (kg/m2) | |
| Mean ±SD | 24.57±2.7 |
| Range | (18.59-29.74) |
| Race | Asian/ 100% |
Pharmacokinetic Parameters
The changes in rivaroxaban plasma concentrations from time 0–48 h post-dose are represented on arithmetic and logarithm scales in Figures 1 and 2, respectively. A non-compartmental analysis was applied for the estimation of PK parameters. Cmax, AUC0-t, AUC0-∞, Tmax, Kel (h−1), T½, of rivaroxaban in plasma concentration are presented in [Table 3].
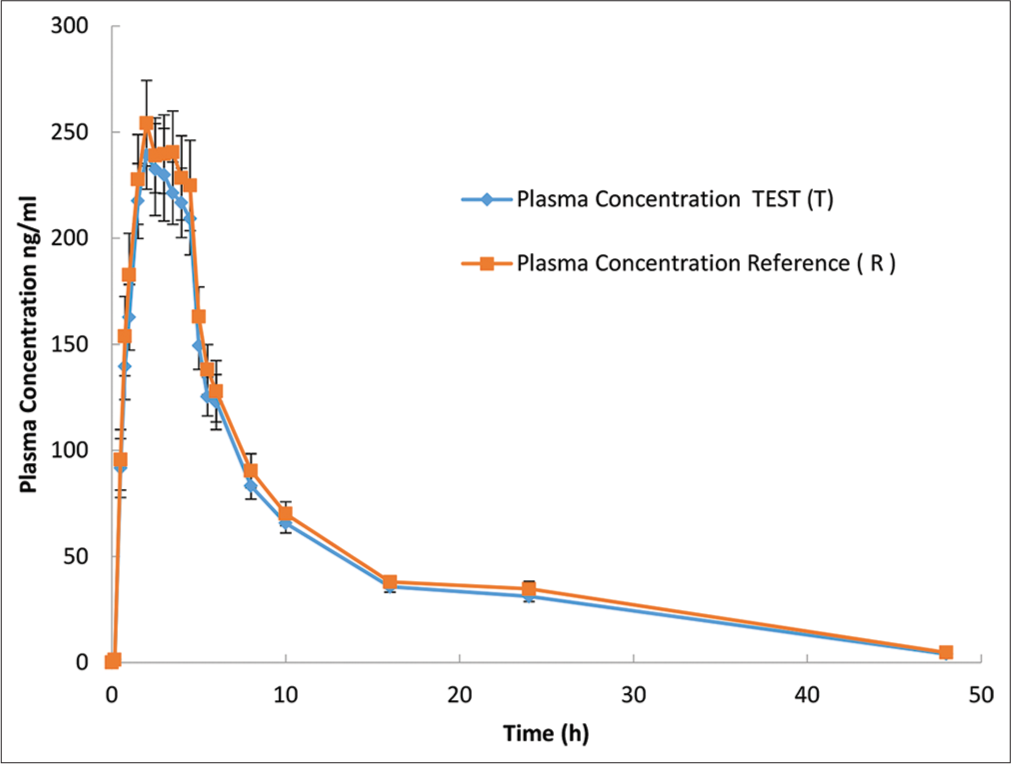
- Rivaroxaban plasma concentration versus time profile for each formulation are presented in an arithmetic scale. Mean (±Standard error) plasma concentration-time of rivaroxaban following a single 20-mg oral dose. Plasma concentration values below the limit of quantification were entered as 0. Blue line indicate a.Asarap® 20 mg (Rivaroxaban Laboratorios Leti S.A.V.), and Red line indicate b. Xarelto 20 mg (Rivaroxaban, Bayer Pvt. Ltd).
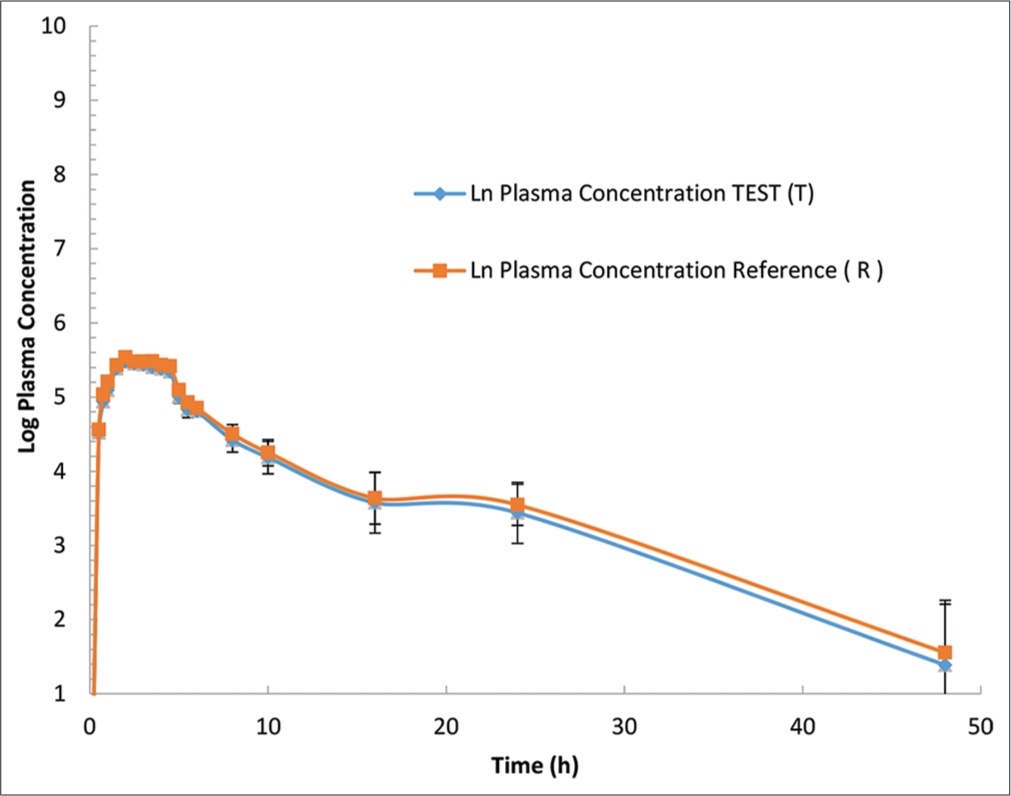
- Rivaroxaban plasma concentration versus time profile for each formulation are presented in a logarithmic scale. Mean (±Standard error) plasma concentration-time of rivaroxaban following a single 20-mg oral dose. Blue line indicate a.Asarap® 20 mg (Rivaroxaban Laboratorios Leti S.A.V.), and Red line indicate b. Xarelto 20 mg (Rivaroxaban, Bayer Pvt. Ltd).
| T (Asarap®) | R (Xarelto®) | |
|---|---|---|
| Cmax(ng/mL) | 279.2±103.4 | 289.4±109.2 |
| AUC0-t(ng*h/mL) | 2345.7±654.0 | 2524.4±872.8 |
| AUC0-∞(ng*h/mL) | 2485.9±660.7 | 2675.4±898.6 |
| ¥Tmax(h) | 2.0 (1.0–4.5) | 2.00 (0.8–4.5) |
| Kel (h−1) | 0.085±0.029 | 0.082±0.026 |
| T1/2(h) | 9.3±4.1 | 9.4±3.7 |
Data presented as a mean±SE. Cmax: Maximum concentration, AUC0-t: Area under the plasma concentration–time curve from time 0 to the last measurable concentration; AUC0-∞: Area under the plasma concentration–time curve from time 0 to infinity, Tmax time to reach Cmax, Kel elimination rate constant, T1/2 time required for plasma concentration to decrease by 50%. ¥Median (range), *Multiplication
All PK parameters calculated for both rivaroxaban formulations after a single dose were similar for both formulations.
Bioequivalence
The test/reference GMRs and the 90% CIs for the logarithm of Cmax, AUC0-t, and AUC0-∞ are in Table 4. They were 87.80% (82.74–93.12), 85.96% (81.88–90.24%), and 86.13% (82.12–90.35%), respectively. These values are within the 90% CI acceptance criteria of 80–125%.
| PK | GMR (T/R) | GMR | 90% CI | |||
|---|---|---|---|---|---|---|
| % | Test (T) | Reference (R) | Lower | Upper | ||
| Cmax(ng/mL) | 87.80 | 252.26 | 287.30 | 82.79 | 93.12 | |
| AUC0-t(ng*h/mL) | 85.96 | 2193.96 | 2552.34 | 81.88 | 90.24 | |
| AUC0-∞, (ng*h/mL) | 86.13 | 2337.67 | 2714.00 | 82.12 | 90.35 | |
Data presented as a % mean Ln transformed. Cmax: Maximum concentration, AUC0-t: Area under the plasma concentration–time curve from time 0 to the last measurable concentration; AUC0-∞: Area under the plasma concentration–time curve from time 0 to infinity. GMR: Geometric mean ratios. n=22. PK: Pharmacokinetic, CI: Confidence interval, Ln: Logarithmic
Study and tolerability
All the 24 subjects were included in the safety assessment. There were no serious and significant effects reported for the study. One subject in the period I reported nausea and one episode of vomiting that lasted for 24 min and it was recorded. Per study protocol, this subject was allowed to complete the study.
DISCUSSION
This aim of this study was to evaluate BE of rivaroxaban 20 mg using as a reference the same dose of the original product Xarelto® by Bayer. The PK mean values of Cmax, AUC0-t, and AUC0-∞ were respectively 279.2 ng/mL, 2345.7 ng*h/mL, and 2485.9 ng*h/mL for T formulation and 289.4 ng/mL, 2524.4 ng*h/mL, and 2675.4 ng*h/mL for R formulation [Tables 5 and 6]. Similar values have been reported for healthy adults under fasting conditions.[18] These similarities in values and shape of the concentration-time curves can also be seen in [Figure 1] where the plasma concentration versus time curves of rivaroxaban for the test and reference formulations are almost overlapping.
Median Tmax was 2.0 h for both the test and the reference [Table 3]. When PK parameters (Cmax, AUC0-t, and AUC0-∞) were analyzed for the T/R ratios, all of them where within the 90% CI BE limits of 80–125% [Table 4].
| Time (h) | Plasma concentration (ng/mL) test (T) | Plasma concentration (ng/mL) reference (R) |
SE (T) | SE (R) |
|---|---|---|---|---|
| 0 | 0.00 | 0.07 | 0.00 | 0.07 |
| 0.167 | 1.34 | 1.32 | 1.00 | 0.84 |
| 0.5 | 91.60 | 95.59 | 13.90 | 14.29 |
| 0.75 | 139.52 | 153.80 | 15.62 | 18.66 |
| 1 | 162.76 | 182.62 | 15.50 | 19.58 |
| 1.5 | 217.50 | 227.68 | 17.65 | 21.21 |
| 2 | 239.28 | 254.15 | 16.24 | 20.14 |
| 2.5 | 232.32 | 239.00 | 21.68 | 17.70 |
| 3 | 229.88 | 239.59 | 21.78 | 18.56 |
| 3.5 | 221.23 | 240.50 | 14.66 | 19.46 |
| 4 | 216.64 | 228.42 | 16.35 | 19.86 |
| 4.5 | 209.11 | 224.83 | 17.00 | 21.27 |
| 5 | 149.30 | 162.97 | 11.19 | 14.07 |
| 5.5 | 125.33 | 138.05 | 9.17 | 11.78 |
| 6 | 122.79 | 127.83 | 12.99 | 14.53 |
| 8 | 83.19 | 90.39 | 6.09 | 8.05 |
| 10 | 65.66 | 70.11 | 4.60 | 5.62 |
| 16 | 35.70 | 37.95 | 2.46 | 2.86 |
| 24 | 31.16 | 34.70 | 2.43 | 3.60 |
| 48 | 4.00 | 4.73 | 1.22 | 1.41 |
Data presented as plasma concentration (mean±SE) per h 0–48 h, in 22 subjects evaluated. SE: Standard error
| Time (h) | Ln plasma concentration (ng/mL) test T (T) | Ln plasma concentration (ng/mL) reference (R) | SE test (T) | SE reference (R) |
|---|---|---|---|---|
| 0 | 0.00 | 0.00 | 0.00 | 0.00 |
| 0.167 | 0.29 | 0.28 | 0.00 | 0.00 |
| 0.5 | 4.52 | 4.56 | 0.07 | 0.07 |
| 0.75 | 4.94 | 5.04 | 0.06 | 0.05 |
| 1 | 5.09 | 5.21 | 0.06 | 0.05 |
| 1.5 | 5.38 | 5.43 | 0.06 | 0.05 |
| 2 | 5.48 | 5.54 | 0.06 | 0.05 |
| 2.5 | 5.45 | 5.48 | 0.05 | 0.06 |
| 3 | 5.44 | 5.48 | 0.05 | 0.05 |
| 3.5 | 5.40 | 5.48 | 0.07 | 0.05 |
| 4 | 5.38 | 5.43 | 0.06 | 0.05 |
| 4.5 | 5.34 | 5.42 | 0.06 | 0.05 |
| 5 | 5.01 | 5.09 | 0.09 | 0.07 |
| 5.5 | 4.83 | 4.93 | 0.11 | 0.08 |
| 6 | 4.81 | 4.85 | 0.08 | 0.07 |
| 8 | 4.42 | 4.50 | 0.16 | 0.12 |
| 10 | 4.18 | 4.25 | 0.22 | 0.18 |
| 16 | 3.58 | 3.64 | 0.41 | 0.35 |
| 24 | 3.44 | 3.55 | 0.41 | 0.28 |
| 48 | 1.39 | 1.55 | 0.82 | 0.71 |
Data presented as Ln plasma concentration (mean±SE) per h 0–48 h, in 22 subjects evaluated. Ln: Logarithmic, SE: Standard error
BE between the test Asarap® and reference product Xarelto® was demonstrated at a dose of 20 mg in this single-dose, crossover study in healthy male subjects under fasting conditions.
The previous studies have also evaluated PK parameters of rivaroxaban 20-mg tablets after a single-dose administration in healthy subjects under fasting conditions[9] and fed conditions[9,18-22] and both fasting condition and fed conditions.[18,22]
Limitations of the study
Rivaroxaban is considered a narrow-therapeutic-index (NTI) drug characterized by low within-subject variability. An acceptable fasting BE study alone does not meet the requirements for approval of rivaroxaban tablets under the guidelines of many regulatory agencies including the United States Food and Drug Administration (US FDA), Health Canada and European Medicines Agency.[22] For example, US FDA recommends that BE be studied on NTI drugs. BE limits the variability of the reference product and compares the within-subject variability of the test and reference products. According to US FDA guidelines, 4-period, 2-sequence, and fully replicated crossover BE studies under fasting and fed conditions are required to assess the within-subject variability of both the test and the reference products of rivaroxaban.[23] Health Canada requires that the 90% CI of T/R ratios be within the range of 90–112% for AUC and 80–120% for Cmax. European Medicines Agency guidelines suggest that the 90%CI of T/R ratios should be within 90–111.11% for both AUC and Cmax.
CONCLUSION
The results of our study demonstrates that test product, Asarap® rivaroxaban 20 mg tablet is bioequivalent and well tolerated as the reference product, Xarelto® rivaroxaban 20 mg tablet in healthy adult male subjects under fasting conditions.
Acknowledgments
This study was conducted at the third party ICBio Clinical Research Pvt. Ltd, located in Vidyaranyapura, Bangalore, India.
Authors contributions
EP, AI and XSM performed the statistical analysis, interpretation, writing and revision of the manuscript.
Declaration of patient consent
The authors certify that they have obtained patient consent.
Conflicts of interest
All authors are Industrias Biocontrolled C.A., (Leti Group Company), employees, and may hold shares and/ or stock options in the company. The authors have no other potential conflicts of interest relevant to this study.
Financial support and sponsorship
This study was funded by Laboratorios Leti S.A.V.
References
- 2022. Available from: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_12
- Trombolismo venoso: Los anticoagulantes orales de acción directa. Rev Fed Arg Cardiol. 2021;46:1-119. 26 de octubre de Avaialble from: https://revistafac.org.ar/ojs/index.php/revistafac/article/view/303
- [Google Scholar]
- Cardiovascular disease burden in the Region of the Americas 2000-2019. ENLACE data portal. In: Pan American Health Organization. 2021.
- [Google Scholar]
- Thrombosis: A major contributor to global disease burden. Arterioscler Thromb Vasc Biol. 2014;34:2363-2371. doi:10.1161/ATVBAHA.114.304488
- [CrossRef] [PubMed] [Google Scholar]
- The mechanism of action of rivaroxaban--an oral, direct Factor Xa inhibitor--compared with other anticoagulants. Thromb Res. 2011;127:497-504. doi:10.1016/j.thromres.2010.09.008
- [CrossRef] [PubMed] [Google Scholar]
- Factor Xa or thrombin: Is factor Xa a better target? J Thromb Haemost. 2007;5(Suppl 1):60-64. doi:10.1111/j.1538-7836.2007.02473.x
- [CrossRef] [PubMed] [Google Scholar]
- Xarelto® (rivaroxaban) Summary of Product Characteristics. 2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/epar_-_product_info [Last accessed on 2022 Dec 04]
- [Google Scholar]
- Pharmacokinetics and Pharmacodynamics of rivaroxaban--an oral, direct factor Xa inhibitor. Curr Clin Pharmacolol. 2014;9:75-83. doi:10.2174/1574884708666131111204658
- [CrossRef] [PubMed] [Google Scholar]
- A single-blind, randomized, single-dose, two-sequence, two-period, crossover study to assess the bioequivalence between two oral tablet formulations of rivaroxaban 20 mg in healthy mexican volunteers. Clin Pharmacol Drug Dev. 2022;11:826-831. doi:10.1002/cpdd.1092
- [CrossRef] [PubMed] [Google Scholar]
- Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin Pharmacol Ther. 2005;78:412-421. doi:10.1007/s40262-013-0100-7
- [CrossRef] [PubMed] [Google Scholar]
- U.S. National Library of Medicine. 2019. Clinical Trials.gov for Bioequivalence Study of Rivaroxaban 10 mg Tablet. Available from: https://clinicaltrials.gov/ct2/show/NCT03071380 [Last accessed on 2022 May 23]
- [Google Scholar]
- Bioavailability and bioequivalence studies In: Pharmaceutical Formulation Design - Recent Practices. London: IntechOpen; 2020. doi: 10.5772/intechopen.85145
- [CrossRef] [Google Scholar]
- ICMR Guidelines 2017. Available from: https://mainicmr.nic.in/sites/default/files/guidelines/ICMR_ethical_guidelines_2017.pdf
- [Google Scholar]
- New drugs and clinical trials rules 2019 India. Perspect Clin Res. 2020;11:37-43. doi:10.4103/picr.PICR_208_19
- [CrossRef] [PubMed] [Google Scholar]
- World Medical Association Declaration of Helsinki, Ethical principles for medical research involving human subjects. Bull World Health Organ. 2001;79:373. doi:10.1001/jama.2013.281053
- [CrossRef] [PubMed] [Google Scholar]
- E6: Note for Guidance on Good Clinical Practice In: PMP/ICH/135/95). Netherlands: European Medicines Agency; 2002.
- [Google Scholar]
- Bioequivalence and food effect assessment of 2 rivaroxaban formulations in healthy Chinese volunteers: An open, randomized, single-dose, and 4-period crossover study. Clinical pharmacology in drug development. Clin Pharmacol Drug Dev. ;2021:11358-363. doi:10.1002/cpdd.103
- [CrossRef] [PubMed] [Google Scholar]
- The effect of food on the absorption and pharmacokinetics of rivaroxaban. Int J Clin Pharmacol Ther. 2013;51:549-561. doi:10.5414/CP201812
- [CrossRef] [PubMed] [Google Scholar]
- Effect of food, an antacid, and the H2 antagonist ranitidine on the absorption of BAY 59-7939 (rivaroxaban), an oral, direct factor Xa inhibitor, in healthy subjects. J Clin Pharmacol. 2006;46:549-558. doi:10.1177/0091270006286904
- [CrossRef] [PubMed] [Google Scholar]
- Impaired rivaroxaban clearance in mild renal insufficiency with verapamil coadministration: Potential implications for bleeding risk and dose selection. J Clin Pharmacol. 2018;58:533-540. doi:10.1002/jcph.1040
- [CrossRef] [PubMed] [Google Scholar]
- Bioequivalence study of 2 formulations of rivaroxaban, a narrow-therapeutic-index drug, in healthy Chinese subjects under fasting and fed conditions. Clin Pharmacol Drug Dev. 2020;9:346-352. doi:10.1002/cpdd.742
- [CrossRef] [PubMed] [Google Scholar]
- FDA Product-Specific Guidances for Generic Drug Development Web Page. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022406.pdf [Last accessed on 2022 Dec 04]
- [Google Scholar]




