
Translate this page into:
Beneficial effects of a polyherbal formulation in the management of sickle cell disease
 , Soliu Abiola Atunwa2, Ismail Ishola3, Lawrence O. Farayola4, Moji Christianah Adeyeye5
, Soliu Abiola Atunwa2, Ismail Ishola3, Lawrence O. Farayola4, Moji Christianah Adeyeye5
*Corresponding author: Adeola Tawakalitu KolaMustapha, PhD Department of Pharmaceutics and Industrial Pharmacy, University of Ilorin, Ilorin, Nigeria. atkmusty@yahoo.com
-
Received: ,
Accepted: ,
How to cite this article: Kola-Mustapha AD, Amali MO, Atunwa SA, Ishola I, Farayola LO, Adeyeye MC. Beneficial effects of a po lyherbal formulation in the management of sickle cell disease. Am J Pharmacother Pharm Sci 2022;6.
Abstract
Objectives:
Inflammation and pain among other comorbid conditions are prominent clinical complications associated with sickle cell disease (SCD). Despite significant improvement in the understanding of SCD pathophysiology, adverse effects of current treatment options are of great concerns. Faradin® (TD) is a polyherbal mixture used in the management of SCD. This study evaluates the acute toxicity, antinociceptive, and anti-inflammatory actions of TD.
Materials and Methods:
Acute toxicity study of TD was conducted according to test guidelines-423 of the Organization for Economic, Cooperation, and Development. Anti-inflammatory effect was assessed with carrageenan-induced paw edema and xylene-induced ear edema while antinociceptive effect was assessed using tail immersion, acetic acid-induced writhing, and formalin-induced nociceptive methods.
Results:
Oral administration of TD showed no acute toxic behavior. TD showed non-dose-related inhibition of inflammation in carrageenan- and xylene-induced edema when compared with vehicle-treated control. Post hoc analysis also revealed that TD caused significant increase in pain threshold in acetic acid, formalin, and tail immersion model of nociception. However, TD-induced antinociception was reversed by naloxone (opioid receptor antagonist) indicative of opioidergic system involvement.
Conclusion:
Findings from this study showed that TD has wide margin of safety and possessed anti-inflammatory as well as antinociceptive properties which lend credence to its potentials in the management of painful and inflammatory conditions associated with SCD.
Keywords
Acute toxicity
Faradin®
Sickle cell disorder
Anti-inflammatory
Antinociceptive
Opioidergic system

INTRODUCTION
Sickle cell disease (SCD) is a life-threatening hematological disorder that affects millions of people worldwide. SCD is caused by lone amino acid substitution in the β-globin chain resulting in polymerization of the abnormal hemoglobin S disrupting erythrocyte architecture and lifespan. Hemoglobin polymerization leads to erythrocyte rigidity, hemolytic anemia, and phases of microvascular vaso-occlusion resulting in organ damage such as the brain, lungs, bone, kidney, and cardiovascular system due to ischemic-reperfusion injury.[1,2] The occurrences of vaso-occlusive events and hemolysis enhance oxidative stress and inflammation.[3] The incidence is estimated to be between 300,000 and 400,000 neonates globally each year, the majority in sub-Saharan Africa.[4] About 18.8% of Nigerian population have the sickle cell trait (SCT; HbAS) which can be genetically transferred to offspring, probably contributing to Nigeria having a high number of people with SCD with about 20–30/1000 live births.[5-7]
Inflammation and excruciating pain are prominent among several clinical complications known to be associated with SCD.[8] Other clinical symptoms are anemia, infections, and diverse complications of vaso-occlusion such as stroke, chest tightening, priapism, leg ulceration, and ultimately chronic organ failure.[9] The recurrent episodes of severe pain which serves as the hallmark of SCD had conferred many names on SCD by different tribes in West Africa. For example, SCD is called Ahotutuo by Twi people of Ghana, rangun-ragun or aromoleegun by the Yorubas of Southwest Nigeria. The pathophysiology of these comorbidities has been attributed to persistent increase in oxidative stress [presence of reactive oxygen species (ROS)], reduction in nitric oxide (NO), and release of inflammatory mediators that cause painful vaso-occlusive crisis (VOC).[10] Regardless of the advancement in the knowledge of the molecular basis of SCD, a standard gold cure is still unavailable. Although some conventional therapies for the management of SCD are available, no universal cure for it exists.[11] Among the drugs recommended to minimize the effects of clinical features are hydroxyurea, 5-hydroxymethyl-2-furfural, hydroxycarbamide,[3] and Sevuparin®.[12] Nonetheless, their untoward effects, for example, secondary leukemia, myelosuppression, mutagenicity, teratogenicity, and inability to completely abolish the complications (VOC) of SCD have limited their usefulness. Other possible therapeutic measures such as hematopoietic cell transplantation,[13] use of endothelins, and blood transfusion,[11] and at times, psychological interventions[14-16] among others have also been explored as therapeutic measures. Unfortunately, their limitations remain high cost, accessibility, and acceptability.[3] It is, therefore, pertinent to discover safe, effective, and affordable anti-sickling agents.
Despite significant development in modern medicine, global extensive use of natural plants as primary health remedies for different disease conditions without the exception of SCD is recognized.[17] A few of the complementary and alternative medicines currently used in Africa for SCD include Niprisan®, Dioscovite®, Hildi®, Ciklavit®, and Faradin®. Faradin® is a polyherbal preparation comprising Zanthoxylum zanthoxyloides (root), Alnus glutinosa (bark), and Alchornea cordifolia (ripe leaves) that have been found anecdotally to be effective for the management of SCD (with no reported adverse effects) in Nigeria and few other West African countries.[18,19] Faradin was approved by the National Agency for Food and Drug Administration and Control (NAFDAC) in Nigeria (Certificate number 4-0077L) as a nutritional supplement. Adeyeye et al. (2017a) and Adeyeye et al. (2017b) established the antibacterial effects of Faradin® (TD) against some Gram-positive and -negative bacteria and reported anti-sickling effects of the extract.[18,19] Several studies have reported antinociceptive action of some of the plants and secondary metabolites in Faradin®.[20,21] It becomes imperative to study the anti-inflammatory and antinociceptive action of this polyherbal formulation in rodents.[22] Hence, establishing the safety profile and validating its anti-inflammatory and antinociceptive activities of the new polyherbal, Faradin® is important in the establishment of its therapeutic benefits in SCD. The objectives of the study are to determine the acute toxicity and evaluate the potential anti-inflammatory and antinociceptive properties of Faradin® using in vivo study in animals.
MATERIALS AND METHODS
Animals
A total number of 135 and 30 Swiss albino mice and Sprague– Dawley rats, respectively, of either sex, were procured from the Animal House of the Department of Biochemistry, University of Ilorin, Ilorin, Nigeria. The Swiss albino mice and rats weigh between 15–20 g and 150–200 g, respectively, and used in this study. The animals were maintained at room temperature under 12 h daylight/night conditions for at least 5 days before the experimental procedures. All the animals had access to water ad libitum and fed with standard diet. Ethical approval number University of Ilorin Ethics and Research Committee (UERC)/ASN/2018/1108 was obtained from the UERC.
Drugs and reagents
Ibuprofen (Hubei Tianyao Pharmaceutical Co. Ltd., China), pentazocine (Sakar Healthcare Ltd., India), diethyl ether, carrageenan 1%, and xylene were procured from Sigma-Aldrich (St. Louis MO, USA). Faradin® was obtained from Atipo Ventures, Ogbomosho, Nigeria.
Experimental procedures
Acute toxicity study
The acute toxicity study was carried out according to the Organization for Economic, Cooperation, and Development (OECD – test guideline 423) up and down procedure.[23] The animals were fasted overnight but had free access to water at least 4 h before the commencement of the study. Fifteen female Swiss albino mice (15–20 g, n = 3) were allotted to three groups comprising vehicle control (10 ml/kg), Faradin® (5, 50, 300, or 2000 mg/kg (volume/weight/kg), p.o.). Then, the animals were observed for behavioral changes such as skin/fur, eyes, mucous, tremor, convulsion, diarrhea, lethargy, sleep, and induction of coma (supplementary file) at 0.5, 1, 2, 4, 6, 8, 12, and 24 h and, subsequently, daily up to the 14th day. Moreover, the influence of treatments on body weight was also recorded for a period of 2 weeks.
Evaluation of anti-inflammatory activity
Carrageenan-induced rat paw edema model
Thirty adult albino rats were fasted and randomized into five groups (n = 6) and treated orally as follows: Vehicle (10 mL/kg) (CTL) (normal control), TD® (25, 50, or 100 mg/kg), and ibuprofen (100 mg/kg) IBU (standard treatment). The paw diameter of the right hind paw was recorded before drug administration. One hour post-drug administration, acute inflammation was induced by subplantar injection of 0.1 mL of 1% suspension of carrageenan in normal saline, in the right hind paw. The paw diameter was measured immediately using a digital Vernier caliper and at 30 min intervals after the carrageenan injection for 4 h. Results were expressed as percentage inhibition of edema and calculated using the formula;[24,25]
Where: C t = Paw thickness at time t; C0 = Paw thickness before administration of treatment and carrageenan; and C t –C0 = difference in paw thickness.
Xylene-induced ear edema model
Another set of adult Albino mice (n = 30) were fasted and randomized into five groups (n = 6). Oral doses were administered to all groups in the order; vehicle (10 mL/kg) labeled CTL, Faradin® (25, 50, or 100 mg/kg) labeled TD25, TD50, and TD100, and ibuprofen (100 mg/kg) labeled IBU. One hour afterward, 30 µL xylene was instilled into the pinna ear of the mouse each to induce edema. The animals were anesthetized with chloral hydrate (300 mg/kg i.p) 30 min later, and both ears were removed and sectioned circularly, using a pair of dissecting scissors. The sections were weighed and percentage inhibition of ear edema was calculated. Results were expressed as percentage inhibition of ear edema and calculated using the formula below.[26,27]
Where: REwt = Right ear weight and LEwt = Left ear weight.
Evaluation of antinociceptive activity
Tail immersion test
Thirty mice were randomly divided into five groups (n = 6) and treated orally; Group 1 – vehicle 10 mL/kg, Groups 2–4: TD (25, 50, or 100 mg/kg, respectively), and Group 5 – morphine 5 mg/kg, s.c. (standard drug). The mouse was restrained in a mouse holder and the tail was immersed in a hot water bath maintained at 52.5 ± 0.5 °C. The reaction time to flick the tail from the hot water was recorded for each mouse. The reaction time was noted initially before any drug treatment and every 30 min for 3 h after the drug treatment. A cutoff time of 15 s was maintained to prevent any injury to the tail. The antinociceptive response was expressed as % maximum possible antinociceptive effect (MPE), which was calculated using a formula;[28]
% MPE = ([Test latency–Control latency] × 100 [Cutoff time–Control latency])
Acetic acid-induced writhing test
Thirty mice were randomly divided into five groups (n = 6) and treated orally as follows; Group 1 – vehicle 10 mL/kg, Groups 2–4: TD (25, 50, or 100 mg/kg, respectively), and Group 5 – ibuprofen 100 mg/kg (positive control). Acetic acid (0.6% v/v, 10 mL/kg) was intraperitoneally administered to mice 1 h post-treatment. Five minutes after acetic acid injection, the total number of writhes (abdominal constriction followed by extension of at least one hind limb) was recorded for 10 min and percentage inhibition was calculated.[26]
Formalin-induced nociceptive behavior
Thirty mice were randomly divided into five groups (n = 6) and treated similar to the arrangement in acetic acid-induced writhing test. One hour post-treatment, formalin 1% v/v, 50 µL was injected into the right hind paw. The time spent in licking and biting the formalin injected paw was recorded for a period of 0–5 min (early phase) and 15–30 min (late phase) considered as the quantitative indication of nociception. The percent inhibition of paw licking time was calculated for early and late phases of nociception in different treatment groups compared with vehicle using the formula:
% inhibition = (CT/C) × 100,
Where: C is the biting/licking response time (s) in the vehicle treatment group and T is the biting/licking response time (s) in the treatment group.[29]
To elucidate the possible mechanism of Faradin-induced antinociception, mice were pre-treated with either naloxone (5 mg/kg, s.c., opioid receptor antagonist), metergoline (5 mg/kg, i.p., 5HT2 receptor antagonist), or prazosin (1 mg/kg, i.p., alpha-1 adrenoceptor antagonist)[25] 15 min before TD or vehicle administration. One hour later, acetic acid-induced mouse writhing test was carried out.
Statistical analysis
Data were expressed as mean ± standard deviation. The results were evaluated using one-way analysis of variance (ANOVA) followed by post hoc test (Tukey’s multiple comparisons test). The GraphPad Prism® version 7.0 was used as the statistical software package.
RESULTS
TD did not produce overt toxic effect
TD at 50, 300, and 2000 mg/kg with the exception of 50 mg/kg induced sedative effect but no observable toxic effect up to 2000 mg/kg for the entire 14 days of treatment. Moreso, there was no significant difference in the mean body weights as compared to the control groups.
TD produced time course decrease in paw edema in carrageenan model
Intraplantar injection of carrageenan 1% w/v, 0.1 mL induced time-dependent increase in paw edema which peaked at 4 h post-injection. However, the pre-treatment of rats with TD (25, 50, or 100 mg/kg, p.o.) significantly (P < 0.05) reduced edema formation with peak effect at 50 mg/kg compared to vehicle-treated control. Moreover, TD inhibited both the early and late phases of inflammation processes. In addition, the anti-inflammatory action of TD at all tested doses was better than the effect of ibuprofen [Figure 1].
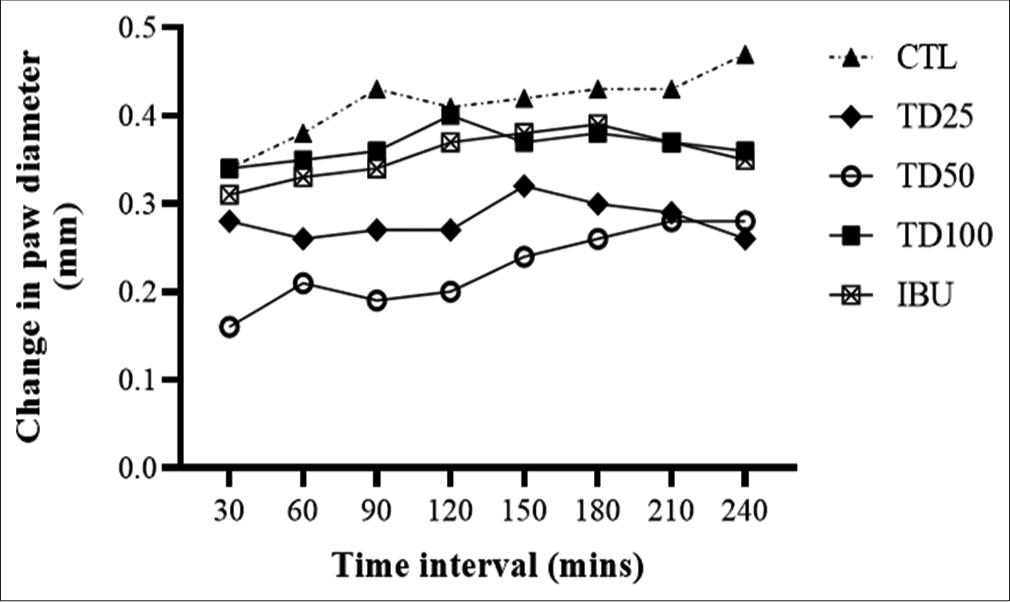
- Effect of TD on carrageenan-induced paw edema in rats. Values are expressed as mean ± SD (n = 6); ap <0.05 versus vehicle-treated control. Statistical level of significance by two-way analysis of variance followed by Tukey’s post hoc multiple comparison test.
TD reduced xylene-induced ear edema
Instillation of xylene into right pinna ear-induced edema formation (42.85% increase) in vehicle-treated control when compared with the left pinna ear. However, the pre-treatment of mice with TD (100 mg/kg) significantly reduced (45.46% inhibition) ear edema formation when compared with vehicle-treated control [Figure 2].

- Effect of TD on xylene-induced ear edema, *P < 0.05 versus vehicle-treated control. Statistical level of significance analyzed by one-way analysis of variance followed by Tukey’s post hoc test of multiple comparison.
TD produced time course increase in supraspinal pain threshold in tail immersion test
Two-way ANOVA revealed significant effect of treatments (F [4,119] = 54.67, P < 0.001). The pre-treatment of mice with TD caused significant time course decrease in nociceptive reaction when compared with vehicle treated, with peak effect at 50 mg/kg but not as potent as morphine (5 mg/kg) [Figure 3].
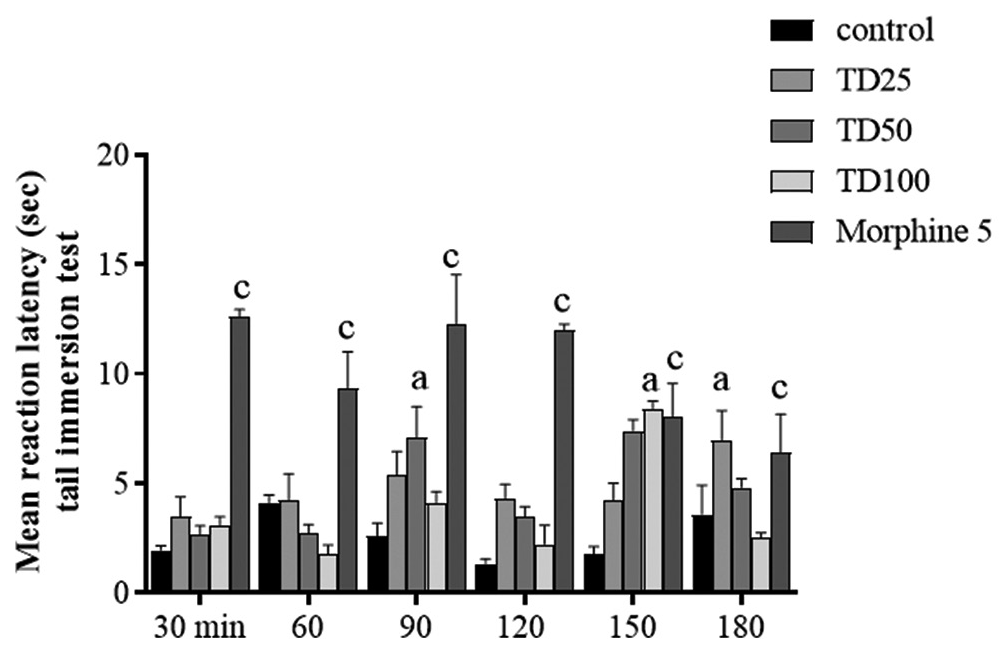
- Effect of TD on hot water tail immersion test. Values are expressed as mean ± SD. Ap < 0.05, cp < 0.001 versus vehicle-treated control, statistical level of significance analysis by two-way analysis of variance followed by Tukey post hoc tests.
TD reduced acetic acid-induced abdominal constriction
Intraperitoneal injection of acetic acid 10 mL/kg induced writhing reflex (21.00 ± 5.61 in 10 min). Writhing nociceptive reaction caused by acetic acid was ameliorated by TD administration with peak effect at TD 50 mg/kg (61.90% inhibition) which was similar to the effect of the standard drug, ibuprofen (78.09% inhibition) in comparison to vehicle-treated control group [Figure 4].
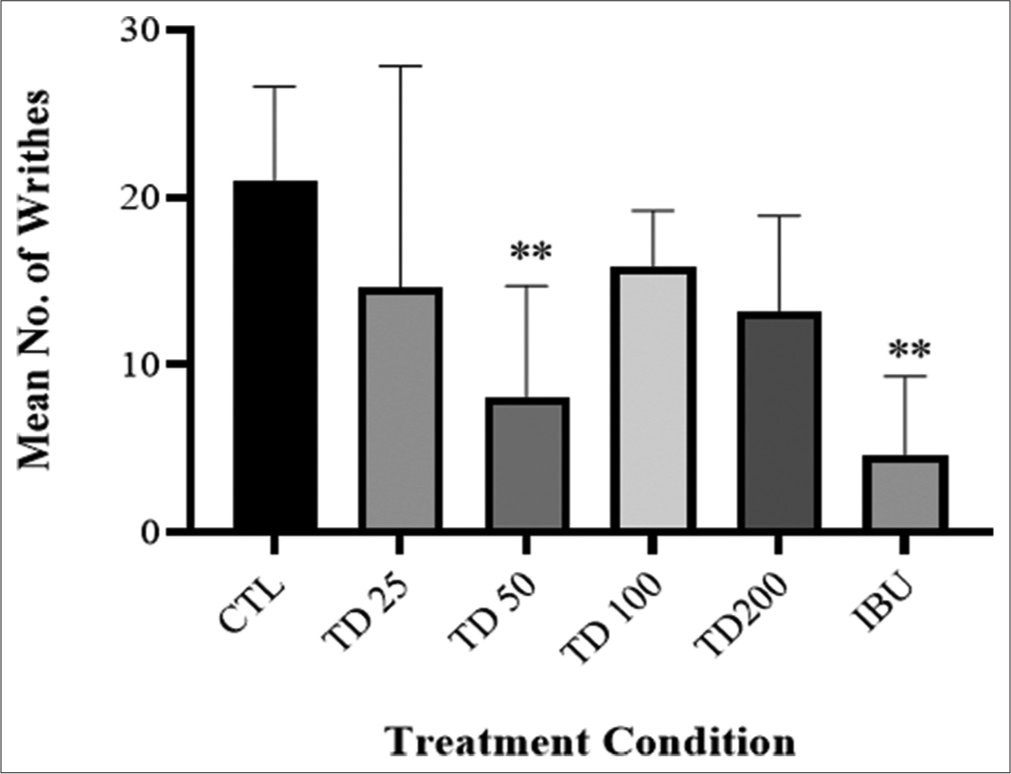
- Effect of TD herbal formulation on acetic acid-induced mouse writhing test. Values are presented as mean ± SD (n = 6). Statistical level of significance analysis by one-way analysis of variance followed by Tukey’s post hoc multiple comparison test, **P < 0.01 versus control.
TD produced biphasic increase in pain threshold in formalin-induced nociception
Intraplantar injection of formalin 1% into the right hind paw-induced biphasic nociceptive behavior including acute early phase (0–5 min) followed by late tonic (15–30 min). TD (50 mg/kg) significantly decreased nociceptive reaction by 62.11% (F [4,40] = 5.40, P < 0.001]) in early phase as well as significant reduction (16.45% inhibition) (F [4,40] = 3.46, P < 0.01]) in time spent licking or biting the injected paw in late tonic phase [Figure 5].

- Duration of paw licking time in formalin-induced nociceptive response. Values are expressed as mean ± SD; *P < 0.01 versus control, statistical level of significance analysis by two-way analysis of variance followed by Tukey’s post hoc tests.
Elucidation of possible mechanism of TD-induced antinociception
Post hoc analysis showed that the subcutaneous administration of naloxone (opioid receptor antagonist) did not affect acetic acid-induced nociception. However, the pre-treatment of mice with naloxone reversed TD-induced antinociceptive action. Moreover, two-way ANOVA revealed significant effect of naloxone and TD treatments (F [1,16] = 6.43, P = 0.02) [Figure 6a]. In another experiment, metergoline (5HT2 receptor antagonist) administration did not modify acetic acid-induced writhing reflex but the pre-treatment of mice with metergoline reduced TD-induced antinociceptive behavior. Two-way ANOVA revealed significant effect of treatments (F [1,16] = 6.84, P < 0.05) and interaction between metergoline and TD treatments (F [1,16] = 13.70, P < 0.05) [Figure 6b]. Post hoc analysis revealed that the pre-administration of prazosin (α1-adrenoceptor antagonist) did not affect mouse writhing reflex but significantly reduced TD-induced antinociceptive behavior. Two-way ANOVA revealed significant effect of prazosin and TD treatments (F [1,16] = 9.29, P < 0.05) and interaction between prazosin/ TD treatments (F [1,16] = 9.68, P < 0.05) [Figure 6c].

- (a-c) Effects of (a) naloxone, (b) metergoline, or (c) prazosin on TD-induced antinociceptive action in mouse writhing assay. Values are expressed as mean ± SD (n = 6). **P < 0.01 versus vehicle-treated control. Statistical level of significance by two-way analysis of variance followed by Tukey’s post hoc multiple comparison test.
DISCUSSION
Findings from this study showed that Faradin® produced anti-inflammatory and antinociceptive activities through inhibition of carrageenan- and xylene-induced edema as well as elevation of pain threshold to both peripheral and central nociception in both acetic acid, formalin, and thermal-induced nociception without affecting body weight and safe up to 2000 mg/kg. The antinociceptive action of Faradin® (TD) was reversed by naloxone (opioid receptor antagonist), metergoline (5HT2 receptor antagonist), and prazosin (alpha-1 adrenergic antagonist) indicative of opioidergic, serotonergic, and adrenergic signaling, respectively.[26,29,30]
Vaso-occlusive crises and intravascular hemolysis have been shown to promote inflammation leading to progressive microvasculopathy.[2] Moreover, vaso-occlusive events lead to activation of sterile inflammation. Carrageenan is a phlogistic agent of choice used in the evaluation of anti-inflammatory agents with no apparent systemic effects.[31] Carrageenan-induced hind paw edema is regarded as a standard experimental model of acute inflammation due to its sensitivity and reproducibility.[25,32] Development of edema in the paw of rat after injection of carrageenan is a discrete biphasic events. The initial phase of which is observed during the 1st h attributed to release of histamine and serotonin, whereas the second phase of edema is due to the release of prostaglandins, protease, and lysosomes.[26] Bhattacharyya et al.[33] showed through an in vitro assay that carrageenan activates Toll-like receptor-4 and reactive oxygen species in human colonocytes similar to what is observe after VOC in SCD. In this study, carrageenan-induced inflammation was reduced by Faradin administration suggestive of its possible benefit in vaso-occlusive crisis.[34] Interestingly, it has been reported that carrageenan-induced edema through stimulation of autacoids release in the 2 h and prostaglandins formation from the 3rd h.[26] In this study, the pre-treatment of rats with Faradin significantly reduced paw edema between the 2nd and 3rd h post-carrageenan injection suggestive of their ability to inhibit autacoids (serotonin, bradykinin, etc.) and prostaglandin formation. In another experiment, the role of Faradin® in the inhibition of inflammatory mediators was assessed in xylene-induced ear edema. Xylene-induced ear edema is an excellent in vivo model for use in evaluating anti-inflammatory potential of new chemical entities.[35] Its action is through release of Substance P from sensory neurons, thereby causing severe vasodilatation, plasma extravasations, and edematous skin changes to the ear of experimental animals.[35] Xylene-induced ear edema assay showed the inhibitory action of Faradin on phospholipase A2, responsible for the breakdown of arachidonic acid into inflammatory markers.[26] This is in tandem with the previous studies,[18,19] where the authors reported the presence of several phytochemicals in the Faradin® extract that has been identified as potential anti-inflammatory agents.
One of the unique features of SCD is recurrent and unpredictable episodes of acute pain due to vaso-occlusive crisis requiring hospitalization. Hence, this study goes ahead to evaluate the antinociceptive effect of Faradin®. The antinociceptive effect of the extract was assessed using three well-validated models, namely, the acetic acid-induced writhing, tail immersion, and formalin-induced nociception tests. This was to determine possible effects of Faradin® against chemical and thermal pain stimuli. The writhing results from intense pain caused by irritation resulting in release of prostaglandins which increased sensitivity to nociceptors, characterized by episodes of contraction of abdominal musculature and stretching of hind limbs over a long period of time similar to what is observed in SCD patients.[25] In this study, Faradin attenuates acetic acid-induced mouse writhing. To further ascertain the peripheral antinociceptive action of Faradin and possible central antinociceptive action, the formalin test was carried out. The formalin test remains the most predictive of the models for acute pain. In this widely used model, a biphasic pain response was produced over a specified test period. Inflammatory phenomena have been observed to occur in between both phases. Some analgesics such as the opioid analgesics have been observed to have both central and peripheral antinociceptive actions. Others such as the NSAIDs seem to inhibit only the second phase.[36-38] Subplantar injection of formalin caused two distinct phases of nociception (licking/biting behavior), initial acute peripheral pain Phase 1 produced through activation of TRPA1 channels, was attenuated by Faradin, recorded in the first 5 min post-injection. Moreover, the second phase (5–15 min post-injection) seen as inflammatory input and central nociception was also inhibited by Faradin. The observed analgesic potential of Faradin® could be attributed to the presence of coumarins, flavonoids, and antioxidants as previously reported.[18] Tail immersion model is a variant of tail flick test and is used in detecting centrally acting analgesic agents with pain induction based on thermal stimulus.[30] It is generally considered to be important for evaluating central analgesic property.[39] In this study, the central antinociceptive action of Faradin was confirmed in tail immersion test evidenced in its ability to increase pain threshold. Moreover, findings from the tail immersion test were corroborated by our mechanistic study, where Faradin-induced antinociception was reversed by naloxone (opioid receptor antagonist) suggestive of opioidergic involvement.
G-protein-coupled receptors such as serotonin, adrenergic, and opioidergic receptors play significant modulatory roles in nociception in the brain areas (amygdala and periaqueductal gray). Moreso, selective serotonin reuptake inhibitors, opioids, and adrenergic agonists relief painful actions in patients. In this study, serotonergic receptors antagonist, metergoline, reversed Faradin-induced antinociception indicative of serotonergic signaling involvement. Interestingly, prazosin (α1 adrenergic receptor antagonist) also blocked Faradin®-induced antinociceptive action suggestive of a role by adrenergic neurotransmission.
Interestingly, the extract up to 2000 mg/kg did not produce any overt behavior of toxicity. All the animals survived up until the 14th day of observation. Thus, Faradin® can be considered to be relatively safe or non-toxic and, therefore, assigned OECD class of Group V. This finding also supports the fact that no claim or report of adverse effect had been filed since Faradin® has been found anecdotally useful in the management of SCD in Nigeria and some West African countries.
CONCLUSION
Findings from this study showed that Faradin® herbal extract possesses anti-inflammatory and analgesic activities through inhibition of possible release of inflammatory markers and increase of pain threshold through adrenergic, serotonergic, and opioidergic signaling. Thus, Faradin® polyherbal mixture may be considered an important remedy in the management of SCD.
Acknowledgment
The authors are grateful to the laboratory staff of the Department of Pharmacology and Toxicology, and College of Medicine for their technical assistance during the study.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
References
- Pathophysiology of sickle cell disease. Ann Rev Pathol. 2019;14:263-292.
- [CrossRef] [PubMed] [Google Scholar]
- Sickle or no Sickle; The Life of the Flesh is in the Blood; No Blood no Life, (Inaugural Lecture Series) Ile-Ife, Nigeria: Obafemi Awolowo University Press; 2019.
- [Google Scholar]
- Sickle-Cell Anaemia: Report by the Secretariat. 2006. Geneva: World Health Organization; Available from: https://www.apps.who.int/iris/handle/10665/20659 [Last accessed on 2019 21 Dec]
- [Google Scholar]
- Sickle cell disease clinical phenotypes in children from South-Western, Nigeria. Niger J Clin Pract. 2015;18:95-101.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence and impact of sickle cell trait on the clinical and laboratory parameters of HIV infected children in Lagos, Nigeria. Pan Afr Med J. 2018;31:113.
- [CrossRef] [Google Scholar]
- Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762-769.
- [CrossRef] [PubMed] [Google Scholar]
- Cytokine profiles in sickle cell anemia: Pathways to be unraveled. Adv Biosci Biotechnol. 2013;4:612.
- [CrossRef] [Google Scholar]
- Sevuparin: Office of Orphan Products Development. 2015. Narrative by Activity Product Designations. United States: Food and Drug Administration; Available from: https://www.fda.gov/media/115531/download [Last accessed on 2019 21 Dec]
- [Google Scholar]
- Hematopoietic cell transplantation for thalassemia and sickle cell disease: Past, present and future. Bone Marrow Transplant. 2008;41:109-117.
- [CrossRef] [PubMed] [Google Scholar]
- Psychosocial therapies for sickle cell disease and pain. Cochrane Database Syst Rev. 2015;8:CD001916.
- [CrossRef] [PubMed] [Google Scholar]
- A cross-cultural study of psychosocial aspects of sickle cell disease in UK and Nigeria. Psychol Health Med. 2007;12:299-304.
- [CrossRef] [PubMed] [Google Scholar]
- Psychosocial impact of sickle cell disorder: Perspective from a Nigerian setting. Global Health. 2014;9:28-34.
- [CrossRef] [Google Scholar]
- Prevalence of use of complementary/alternative medicine: A systematic review. Bull World Health Organ. 2000;78:252-257.
- [Google Scholar]
- Evaluation of an undocumented polyherbal (Faradin®) used for the treatment of sickle cell disease in West Africa. Part I: Phytochemistry and ex-vivo anti-sickling study. J Pharm Res Int. 2017a;17:1-14.
- [CrossRef] [Google Scholar]
- Evaluation of an undocumented polyherbal (Faradin®) used for the treatment of sickle cell disease in West Africa. Part II: Antibacterial activity and synergism. J Pharm Res Int. 2017;17:1-11.
- [CrossRef] [Google Scholar]
- A Modern Herbal: Alder. E-book Library. 2004. Available from: https://www.botanical.com/botanical/mgmh/a/alder019.html [Last accessed on 2019 Dec 21]
- [Google Scholar]
- Medicinal and Economic Plants of Nupe Land (1st ed). Bida, Nigeria: Jube-Evans Books and Publications; 2003. p. :106-107.
- [Google Scholar]
- Capacity for clinical research on herbal medicines in Africa. J Alternat Complement Med. 2012;18:622-628.
- [CrossRef] [PubMed] [Google Scholar]
- OECD Guidelines for the Testing of Chemicals/Section 4: Health Effects Test No. 423. In: Acute Oral Toxicity-Acute Toxic Class Method. France: Organization for Economic Cooperation and Development; 1996.
- [Google Scholar]
- Analgesic and anti-inflammatory effect of the aqueous extract of leaves of Persea americana (Lauraceae) Fitoterapia. 2002;73:375-380.
- [CrossRef] [Google Scholar]
- Antinociceptive and anti-arthritic properties of hydroethanolic leaf extract of Clausena anisata (Willd) Hook. f. ex Benth (Rutaceae) in rodents: Possible mechanism of actions. Niger J Physiol Sci. 2015;30:39-49.
- [Google Scholar]
- Analgesic and anti-inflammatory activities of Cnestis ferruginea Vahl ex DC (Connaraceae) methanolic root extract. J Ethnopharmacol. 2011;135:55-62.
- [CrossRef] [PubMed] [Google Scholar]
- Phytochemical and anti-inflammatory studies of ethanol extract of Terminalia macroptera Guill and Perr (Combretaceae) stem bark in rats and mice. Niger J Pharm Res. 2018;13:147-156.
- [Google Scholar]
- Antinociceptive effect of certain dimethoxy flavones in mice. Eur J Pharmacol. 2014;727:148-157.
- [CrossRef] [PubMed] [Google Scholar]
- Antinociceptive, anti-inflammatory and antiulcerogenic activities of ethanol root extract of Strophanthus hispidus DC (Apocynaceae) J Basic Clin Physiol Pharmacol. 2013;24:277-286.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms of analgesic and anti-inflammatory properties of Annona muricata Linn (Annonaceae) fruit extract in rodents. J Med Food. 2014;17:1375-1382.
- [CrossRef] [PubMed] [Google Scholar]
- In-vivo anti-inflammatory activities of leaf extracts of Ocimum lamiifoliumin mice model. J Ethnopharmacol. 2011;134:2-36.
- [CrossRef] [PubMed] [Google Scholar]
- Studies on the mediators of the acute inflammatory response induced in rats in different sites by carrageenan and turpentine. J Pathol. 1971;104:15-29.
- [CrossRef] [PubMed] [Google Scholar]
- Toll-like receptor 4 mediates induction of the Bcl10-NFkappaB-interleukin-8 inflammatory pathway by carrageenan in human intestinal epithelial cells. J Biol Chem. 2008;283:10550-10558.
- [CrossRef] [PubMed] [Google Scholar]
- Carrageenan-induced inflammation promotes ROS generation and neutrophil extracellular trap formation in a mouse model of peritonitis. Eur J Immunol. 2016;46:964-970.
- [CrossRef] [PubMed] [Google Scholar]
- Antinociceptive and anti-inflammatory properties of Tetracera alnifolia Willd (Dilleniaceae) hydroethanolic leaf extract. J Basic Clin Physiol Pharmacol. 2018;30:173-184.
- [CrossRef] [PubMed] [Google Scholar]
- The formalin test: A quantitative study of the analgesic effects of morphine, merperidine and brain stem stimulation in rats and cats. Pain. 1977;4:161-174.
- [CrossRef] [Google Scholar]
- A new method of pain scoring in the formalin test in rats. Pain. 1997;71:265-270.
- [CrossRef] [Google Scholar]
- Pharmacological effect and toxicity of alkaloids from Gelsemium elegans (Benth) J Ethnopharmacol. 2003;89:91-95.
- [CrossRef] [Google Scholar]




