
Translate this page into:
Bioequivalence assessment of two formulations of empagliflozin in healthy adult subjects
*Corresponding author: Evelyn Pena, MSc MD Medical Department Industrias Biocontrolled C.A. Leti S.A.V., Guarenas, Miranda, Venezuela. martinpena24@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Pena E, Inatti A, Taly A, et al. Bioequivalence assessment of two formulations of empagliflozin in healthy adult subjects. Am J Pharmacother Pharm Sci 2023;019.
Abstract
Objective:
The objective of the study was to evaluate the bioequivalence (BE) between Izaban® (test) and Jardiance® (reference) empagliflozin 25 mg, oral tablets, in healthy adult subjects.
Materials and Methods:
A randomized, open-label, two-sequence, and two-period crossover comparative oral bioavailability study was conducted on healthy adult subjects. It was tested BE in vivo using a comparative pharmacokinetic (PK) evaluation. Serial blood samples were collected up to 72 h following oral administration of the study drugs, plasma concentrations of empagliflozin were using liquid chromatography mass spectrometry (LC-MS-MS) method.
Results:
The test and reference drug products were considered bioequivalent when the geometric means of the test (T)/reference (R) ratios and 90% confidence intervals (CIs) fall within the range of 80.00–125.00%. For PK parameters, % T/R ratios and 90% CIs were Cmax: 105.11% (100.28–110.18%), area under curve (AUC0-t): 103.25% (99.62–107.00%), and AUC0-∞ 102.71% (99.26–106.28%).
Conclusion:
Our study demonstrated in vivo BE between the two empagliflozin formulations tested in healthy subjects under fasting conditions.
Keywords
Empagliflozin
Bioequivalence
Pharmacokinetics

INTRODUCTION
The pharmacological management of type 2 diabetes mellitus (T2DM) involves use of new therapies for glycemic control and prevention of cardiovascular complications of diabetes while decreasing morbidity and mortality.[1] Uncontrolled diabetes can lead to a number of short- and long-term complications, including heart disease, dyslipidemia, diabetic nephropathy, and retinopathy.[1] Therefore, to prevent the development of diabetic complications, more effective therapies should be used to manage blood glucose control. Sodium glucose cotransporter-2 inhibitors (SGLT2i) are new oral drugs for the therapy of patients with (T2DM). The major pharmacological action of SGLT2i is through inhibiting glucose reabsorption in the kidney and thus promoting glucose excretion.[2,3]
Empagliflozin was the first SGLT2i approved, for the treatment of adults with T2D globally under the brand name, Jardiance® Empagliflozin enhances renal glucose excretion or glycosuria and reduces hyperglycemia in an insulin-independent manner by highly selective inhibition of SGLT2.[4]
The expression of SGLT2 is up-regulated, and the urinary glucose excretion threshold is also higher in patients with hyperglycemia compared with healthy humans.[3,5] Inhibition of SGLT2 reduces glucose reabsorption, promotes urinary glucose excretion, and produces negative caloric balance, which leads to weight loss.[1,5] SGLT2i can thus be used on top of other oral glucose-lowering drugs and insulin to exert additive anti-hyperglycemic effects.[2-4]
The recommended starting dosage of empagliflozin is 10 mg once daily. The dosage may be increased to a maximum of 25 mg/day in patients tolerating empagliflozin 10 mg/day.[5,6]
The pharmacokinetic (PK) profile after oral administration, peak plasma concentrations (Cmax) of empagliflozin were reached at 1.33–3.0 h post-dose.[1,5,6] Thereafter, plasma concentrations declined in a biphasic manner with a rapid distribution phase and a relatively slow terminal phase. The steady state mean plasma the area under curve (AUC) and Cmax were 1870 nmol*h/L and 259 nmol/L, respectively, with 10 mg empagliflozin once daily treatment, and 4740 nmol*h/L and 687 nmol/L, respectively, with 25 mg empagliflozin once daily treatment. Systemic exposure of empagliflozin increased in a dose proportional manner in the therapeutic dose range.[1,5,6] The single-dose and steady-state PK parameters of empagliflozin were similar, suggesting linear PKs with respect to time. Administration of 25 mg empagliflozin after intake of a high-fat and high-calorie meal resulted in decrease in AUC by approximately 16% and Cmax d by approximately 37%, compared to fasting state.[1,5,6] The observed effect of food on empagliflozin PKs was not considered clinically relevant and empagliflozin may be administered with or without food.[1,5,6] The apparent steady-state volume of distribution was estimated to be 73.8 L based on a population PK analysis and plasma protein binding was 86.2%.[1,5,6] No major metabolites of empagliflozin were detected in human plasma and the most abundant metabolites were three glucuronide conjugates (2-O-, 3-O-, and 6-O-glucuronide). Systemic exposure of each metabolite was <10% of total drug-related material. The route of metabolism of empagliflozin in humans is glucuronidation by the uridine 5’-diphospho-glucuronosyltransferases UGT2B7, UGT1A3, UGT1A8, and UGT1A9.[1,2,5] The apparent terminal elimination half-life of empagliflozin was estimated to be 12.4 h and apparent oral clearance was 10.6 L/h based on the population PK analysis. Following once-daily dosing, up to 22% accumulation, with respect to plasma AUC, was observed at steady-state, which was consistent with empagliflozin half-life (V1/2). Approximately 95.6% of the drug-related radioactivity was eliminated in feces (41.2%) or urine (54.4%).[1,5]
The purpose of our study was to assess and compare the PK profiles and safety of Jardiance® (Boehringer Ingelheim Pharmaceuticals, Inc.) as a reference to Izaban® (Laboratorios Leti, S.A.V., República Bolivariana de Venezuela) as a test (T) formulation of empagliflozin 25 mg, in healthy adult subjects under fasting conditions.[7] This study was conducted by CRO ICBio Clinical Research Pvt, Ltd. India.
MATERIALS AND METHODS
Ethical approval
The study was conducted ethically in accordance with the principles of the ICMR guidelines (2017),[8] New Drugs and Clinical Trials Rules 2019 India,[9] and adhered to the ethical principles of the Declaration of Helsinki,[10] the International Conference on Harmonization Good Clinical Practice Guidelines.[11]
The study protocol was approved by an Independent Ethical Committee (ECR/141/indt/KA/2013/RR-19), Application N°EC/RENEW/IND2019/6255 and certified by CDSCO/ DGHS to ICBio Clinical Research Pvt, Ltd. Study number: ICBio/029/0722.
Study design
This was an open-label, randomized, two-treatment, two-period, two-sequence, single oral dose, and crossover bioequivalence (BE) study under fasting conditions comparing, two empagliflozin formulations.
Izaban® 25 mg tablets were provided as the test formulation (T) by Laboratorios Leti S.A.V, República Bolivariana de Venezuela, batch N°EP-0322528-4E, date of expiry May 2024, and the reference formulation (R) Jardiance® 25mg tablets of Boehringer Ingelheim Pharmaceuticals, Inc., Germany, batch N°E02292, date of expiry 05–2025.
According to randomization schedule, a single dose of the study drug (T or R) was administered in each period, as shown in [Table 1]. Subjects who received T product in period I were administered R product in period II and vice versa. Pre-screening period was 21 days. The study lasted for 15 days (January 04, 23–January 17, 23) with 12 day washout period considering the terminal half-life for empagliflozin is 12.5 h.[1,5,6]
| Sub. No: | Sequence | Period I | Period II |
|---|---|---|---|
| 1 | TR | T | R |
| 2 | RT | R | T |
| 3 | RT | R | T |
| 4 | TR | T | R* |
| 5 | RT | R | T |
| 6 | TR | T | R |
| 7 | TR | T | R |
| 8 | RT | R | T |
| 9 | RT | R | T |
| 10 | TR | T | R |
| 11 | TR | T | R |
| 12 | TR | T | R |
| 13 | TR | T | R |
| 14 | TR | T | R |
| 15 | RT | R | T |
| 16 | TR | T | R |
| 17 | TR | T | R |
| 18 | RT | R | T |
| 19 | RT | R | T |
| 20 | RT | R | T |
| 21 | RT | R | T |
| 22 | RT | R | T |
| 23 | RT | R | T |
| 24 | TR | T | R |
R: Reference, Jardiance®, T: Test, Izaban®. (*) The subject 04 did not show up for the second period and was considered a dropout
Subjects
Twenty-four male volunteers aged between 18 and 45 years participated in the study. They had a mean age 31.83 ± 5.6 years, mean weight 70.63 ± 8.4 Kg, mean height 1.68 ± 0.04 meters, and body mass index of 25.02 ± 2.6 kg/m2 [Table 2].
| Data of subjects | Treatment groups | |
|---|---|---|
| Test product n=24 | Reference product n=23 | |
| Age (years) | ||
| Mean±SD | 31.83±5.6 | 50±15 |
| Range 18–40 |
20–40 | 20–40 |
| Age groups | ||
| 18–40 | 24 (100%) | 23 (100%) |
| Weight (kg) | ||
| Mean±SD | 70.63±8.4 | 70.03±8.4 |
| Range | 57–84 | 57–84 |
| Height (m) | ||
| Mean±SD | 1.68±0.04 | 1.67±0.02 |
| Range | 1.60–1.79 | 1.60–1.79 |
| BMI (kg/m2) | ||
| Mean±SD | 25.02±2.69 | 24.99±2.03 |
| Range | 19.72–29.41 | 19.72–29.41 |
| Race | ||
| Asian | 24 (100%) | 23 (100%) |
| Sex M/F | ||
| Male | 24 (100%) | 23 (100%) |
BE: Bioequivalence, BMI: Body mass index, SD: Standard deviation
Only male subjects fulfilled all the following inclusion criteria and were accepted to start this study because the empagliflozin PK is not affected by age, gender, body mass index, or race.[1,5,6] A complete clinical history valid for 6 months before the start of the study; normal laboratory values as determined by medical history and physical examination at the time of screening; normal vital signs and physical examination; creatinine clearance of more than 50 mL/min; negative tests for hepatic transaminases, hepatitis B and C, human immunodeficiency virus, and venereal diseases research laboratory; and normal 12-lead EKG values, normal chest radiography, and negative result in urine drug tests. Other key inclusion criterion is that subjects must be non-smokers or smokers who had not smoked at least 10 h before the start of the study. All the subjects were informed about the possible risks and the benefits of their participation such as blood laboratory test, electrocardiogram, as well as travel and food expenses. They all signed the informed consent.
The exclusion criteria included a history of hypersensitivity to the study medication or to any other medication belonging to the study group or cardiovascular, renal, hepatic, metabolic, gastrointestinal, neurological, endocrine, hematopoietic, psychiatric, or other organic abnormalities; under medication that interferes with the quantification and/or kinetics of the medication under study or potentially toxic medications within 30 days before the start of the study; exposure to agents known as inducers or inhibitors of liver enzyme systems; taken any medication, hospitalized for any reason or who were seriously ill within the 90 days before the study; donated or lost 300 mL or more of blood within 90 days before the start of the study; recent history of drug abuse, including alcohol; consumed products such as cola drinks containing caffeine, theobromine, or theophylline in the 48 h before the study; and grapefruit juice consumption in the 72 h before the study.
Drug administration and blood collection
Following an overnight fast, subjects were scheduled for dosing per the randomization schedule in each period [Table 1]. All the subjects fasted for at least 10 h pre-dose and 4 h post-dose. Single dose of either one T or R was administered with 240 mL of 20% glucose solution at ambient temperature, followed by 60 mL the 20% glucose every 15 min for up to 4 h after dose of T or R product.[7] All subjects were in sitting posture for 02 h after dosing.[7,12-16]
The subjects received standardized meals at 04.00, 08.00, 12.00, and 24.00 h after dosing in each period. During housing, the meal menu was same in both the periods (2500 Kcal) and drinking water was provided ad libitum.
The study was conducted with 24 subjects for period I and 23 subjects for period II. One subject (patient 04) did not show up for the second period and was considered a dropout. A total of 20 × 6 mL blood samples were collected through cannula from each subject during each period, while 24:00, 48:00, and 72 h samples were collected by direct venipuncture. The venous blood samples were withdrawn at pre-dose (00.00 h) and 00.33, 00.66, 01.00, 01.25, 01.50, 01.75, 02.00, 02.50, 03.00, 03.50, 04.00, 05.00, 06.00, 08.00, 10.00, 12.00, 24.00, 48.00, and 72.00 h, post-dose. Equal allocation of treatments or balanced randomization is shown [Table 1].
Analytical methodology
The blood samples were collected in pre-labeled K2 and ethylenediaminetetraacetic acid (EDTA) vacutainers and were centrifuged at 4000 rpm for 10 min at 2–8°C, plasma was separated, labeled, and stored at −70°C ± 5°C before analysis. Subsequently, the plasma samples were processed, calibration curve of internal standards (IS) Empaglifozin D4, (Vivian Life Sciences Private Limited, Mumbai, India) and quality control (QC) samples were thawed and vortexed for preparation and analysis. Empagliflozin was selectively isolated from 300 µL plasma by liquid–liquid extraction method. Aliquots of 0.300 µL were mixed with 0.300 µL of extraction buffer (50 mM sodium carbonate in water) and vortexed. After was add 2.500 mL of TBME (methyl tert-butyl ether) and shaken for 10 min, the samples were centrifuged at 4000 rpm for 5 min at 4°C. A 2.000 mL of the supernatant layer were transferred and dried in nitrogen evaporator at 40°C ± 2°C. The samples were reconstituted with 0.50 mL of reconstitution solution and spiked with IS over the concentration range of 4.809–600.531 ng/mL. Analytes, IS, and QC samples were transferred to pre-labeled vials in the autosampler at 5°C ± 3°C and injected in an liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS-MS) instrument (Shimadzu LCMS-8040 Mumbai, India). The chromatographic separation was performed by a BDS Hypersil C18, 4.6 * 00 mm, id 5 µm high performance liquid chromatography (HPLC) column (Thermo Scientific, Mumbai, India). The mass spectrometer was operated in positive electrospray mode. Identifications were based on multiple reactions monitoring transitions; m /z 472.05–359.00 for empagliflozin and m/z 468.15–355.10 for IS. The inter-batch calibration standard accuracy was 93.6–104.27% with precision values of 2.13–4.57% and acceptable linearity.
Statistical analysis of PKs parameters for BE determination
The sample size calculation for the study was based on intra-subject coefficient of variation (CV%) for empagliflozin as obtained from published literature (Cmax14–25%, AUC0-∞16–24%).[5,6,12-16]
With the expected % CV for Cmax and AUC not exceeding 20% and the ratio between within 95 and 105% (i.e., a true treatment difference of 5%), the study required 20 evaluable subjects to demonstrate BE with a power of 90% at 5% level of significance. Additional subjects were included in the study for possible dropouts/withdrawals. Thus, a total of 24 healthy subjects were sufficient to demonstrate BE between the T and R products. The PK parameters calculated were maximum peak concentration (Cmax), AUC from time 0 h to the last measurable concentration (AUC0-t), AUC from time 0 to infinity (AUC0-∞), and time to reach Cmax (Tmax) as primary parameters to PK analysis. Others secondary PK parameters evaluated were as follows: AUC _% Extrap_obs, Tmax, T1/2, and λz/Kel of empagliflozin in plasma. PK and statistical analyses were performed using SAS version 9.3.1 Inc., Cary, North Carolina. USA. The log-transformed PK parameters were analyzed using a general linear model (Proc GLM of SAS® Mumbai, India). The formulations were regarded as bioequivalent when the 90% confidence intervals (CIs) of the T and R ratio of AUC0-t, AUC0-∞ and Cmax ranged from 80 to 125%.[7] This is the standard accepted by the United States Food and Drug Administration, (FDA).[7,12-16]
Safety assessment
The safety of two formulations was evaluated through the assessment of adverse events (AEs) monitoring throughout the study. According FDA rules, subjects were a test or parent reference formula in 240 mL of 20% glucose solution and 60 mL of 20% glucose solution every 15 min for 4 h after dosing to reduce the risk of hypoglycemia, while fasting empagliflozin BE analysis.[7] All AEs were recorded based on the Medical Dictionary for Regulatory Activities. The Common Terminology Criteria for AEs were used for assessment of severity. Vital signs were measured during baseline screening, and at the conclusion of the study. Twelve-lead electrocardiogram and clinical laboratory such as urine analysis, hematology, and blood biochemistry including glycemia evaluation were conducted during screening and again 72 h after the study.
RESULTS
PKs parameters
PK and statistical analysis were performed for 23 subjects (subject 04 was a drop out, did not attend the period II) and parameters obtained from subjects following oral dosing of empagliflozin 25 mg for 72 h post-dose are represented on arithmetic and logarithm scales, as shown in [Figures 1 and 2]. A non-compartmental analysis was applied for the estimation of PK parameters Cmax, AUC0-t, AUC0-∞ Tmax, Kel (h−1), and T½, of empagliflozin in plasma concentration which are presented in [Table 3], analysis of variance analysis from Ln Cmax, AUC0-t, and Ln AUC0-∞. There were no significant differences between the PK parameters of the two empagliflozin formulations (P > 0.05).
| PK Parameters | Mean±SD | |
|---|---|---|
| Test (T) | Reference (R) | |
| Cmax(ng/mL) | 357.28±102.70 | 339.22±92.14 |
| AUC0-t(ng*h/mL) | 3256.41±877.08 | 3136.4574±741.32 |
| AUC0-∞(ng*h/mL) | 3522.8373±885.92 | 3409.2551±716.71756 |
| Tmax(h)# | 3.000 (1.5–5.0) | 3.500 (1.500–5.000) |
| Kel(h-1) | 0.0958±0.0166 | 0.0943±0.0127 |
| T1/2(h) | 7.44±1.31 | 7.47±0.97 |
Data presented as a mean±standard error. Cmax: Maximum concentration, AUC0-t: Area under the plasma concentration–time curve from time 0 to the last measurable concentration, AUC0-∞: Area under the plasma concentration–time curve from time 0 to infinity, Kel: Elimination rate constant, Tmax: Time to reach Cmax, T1/2time required for plasma concentration to decrease by 50%, PK: Pharmacokinetic
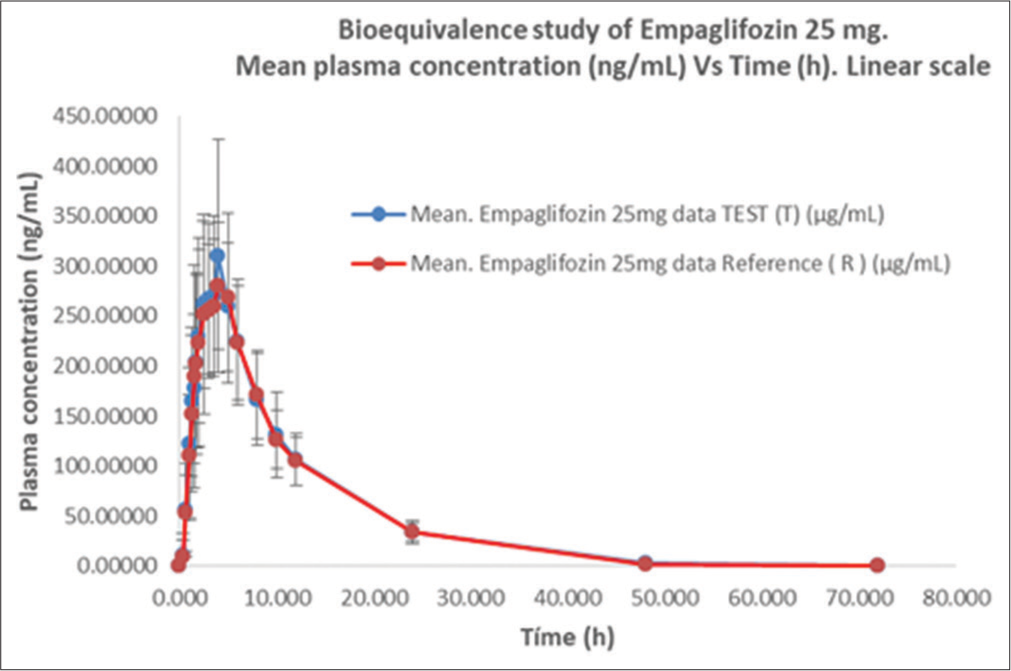
- Empagliflozin plasma concentration versus time profile for test (T) and reference (R) formulations, (mean ± SD) following a single 25 mg oral dose. Arithmetic scale
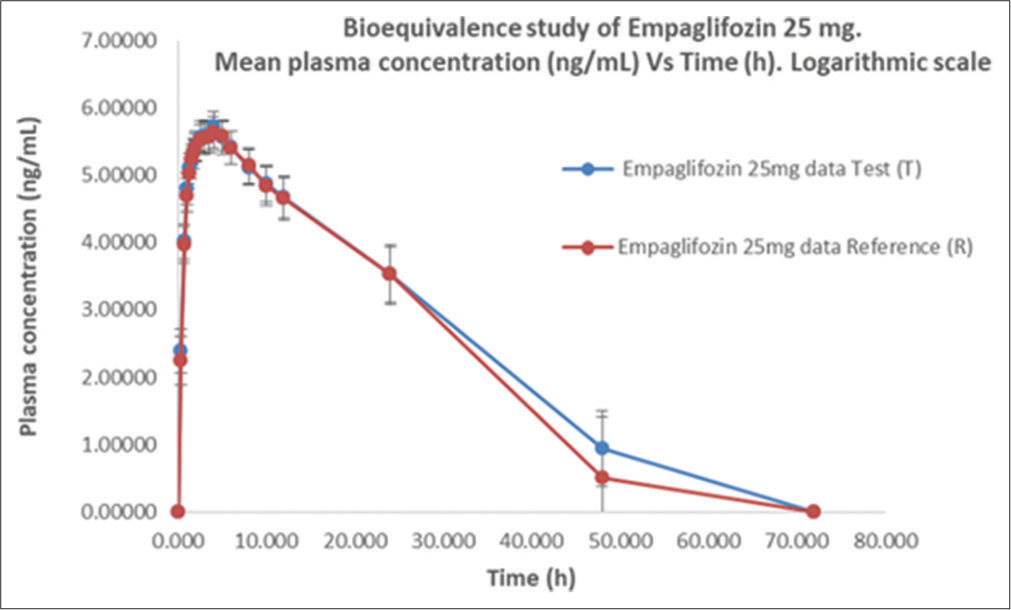
- Empagliflozin plasma concentration versus time profile for test (T) and reference (R) formulations, (mean ±SD) following a single 25 mg oral dose. Logarithmic scale.
The test/reference geometric mean ratios and 90% CIs for the logarithm of Cmax, AUC0-t, and AUC0-∞ are presented in [Table 4]. The BE results were as follows: Ln Cmax 105.11% (CI 100.28–110.18%), AUC0-t 103.25% (CIs 99.62–107.00%), and Ln AUC0-∞ 102.71% (CI 99.26–106.28%); these values are within the 90% CIs, acceptance criteria of 80–125% [Table 4].[7,12,15]
| PK parameters | GMR ratio % (test/ref) | 90% CI | |
|---|---|---|---|
| Lower limit | Upper Limit | ||
| Cmax(ng/mL) | 105.11 | 100.28 | 110.18 |
| AUC0-t(ng*h/mL) | 103.25 | 99.62 | 107.00 |
| AUC0-∞(ng*h/mL) | 102.71 | 99.26 | 106.28 |
Data presented as a % mean Ln transformed. Cmax: maximum concentration, AUC0-t: Area under the plasma concentration–time curve from time 0 to the last measurable concentration, AUC0-∞: Area under the plasma concentration–time curve from time 0 to infinity. BE acceptance criteria of 80–125%, GMR: Geometric mean ratios, CI: Confidence interval, PK: Pharmacokinetic
Safety results
Subjects were monitored for their well-being by recording vital signs before, during, and at the end of study. Blood glucose level (random blood glucose) ware checked pre-dose, 03.00 and 06.00 h post-dose or required for the safety of the subjects. Post-study safety evaluation was performed on all subjects who completed the period II. On evaluation, all the safety parameters were found to be normal. No severe, serious, or life-threatening side effects were reported throughout the study.
DISCUSSION
The initial commercialization of Jardiance® by Boehringer Ingelheim Pharmaceuticals, Inc., introduces the first formulation of empagliflozin in 5, 12.5, and 25 mg doses.
This study aimed to compare the PK profile and safety of 25 mg of two formulations in fasting condition based on US FDA Guidance on Empagliflozin BE studies.[7] The PK mean values of Cmax, AUC0-t, and AUC0-∞ were, respectively, 357.20 ng/mL, 3256.41 ng*h/mL, and 3522.83 ng*h/mL for T and 339.22 ng/mL, 3136.45 ng*h/mL, and 3409.25 ng*h/mL for R. These parameters was evaluated, in fasting conditions with a washout period of 12 days, which covered >5 times the half–life of empagliflozin. Others secondary PK parameters were similar, as shown in [Table 3], the mean to Tmax was 3.0 h and 3.5 h and T1/2 was 7.44 h and 7.47 h to T and R, respectively. These similarities in values and shape on the concentration–time curve are shown in [Figures 1 and 2, Table 5]. The 90% CIs of Cmax, AUC0-t, and AUC0-∞ for empagliflozin are falling within the BE acceptance range of 80.00–125.00%.[7,12-16]
| Time (h) | Mean. Empaglifozin 25mg data test (T) (µg/mL) |
Empaglifozin 25 mg data log test (T) | SD data test (T) | Mean. Empaglifozin 25 mg data reference (R) (µg/mL) |
Empaglifozin 25 mg data log reference (R) | SD data reference (R) |
|---|---|---|---|---|---|---|
| 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 0.334 | 10.83 | 2.38 | 0.32 | 9.48 | 2.24 | 0.35 |
| 0.667 | 55.49 | 4.01 | 0.26 | 52.86 | 3.96 | 0.27 |
| 1.00 | 121.98 | 4.80 | 0.23 | 110.04 | 4.70 | 0.24 |
| 1.25 | 164.49 | 5.10 | 0.23 | 152.05 | 5.02 | 0.22 |
| 1.50 | 177.35 | 5.17 | 0.30 | 189.11 | 5.24 | 0.21 |
| 1.75 | 204.25 | 5.31 | 0.22 | 202.51 | 5.31 | 0.22 |
| 2.00 | 229.54 | 5.43 | 0.22 | 223.75 | 5.41 | 0.21 |
| 2.50 | 261.81 | 5.56 | 0.22 | 252.06 | 5.52 | 0.21 |
| 3.00 | 267.27 | 5.58 | 0.23 | 255.18 | 5.54 | 0.23 |
| 3.50 | 270.18 | 5.59 | 0.22 | 259.92 | 5.56 | 0.23 |
| 4.00 | 309.59 | 5.73 | 0.21 | 280.36 | 5.63 | 0.24 |
| 5.00 | 259.14 | 5.55 | 0.24 | 268.21 | 5.59 | 0.22 |
| 6.00 | 224.03 | 5.41 | 0.24 | 223.52 | 5.40 | 0.24 |
| 8.00 | 166.37 | 5.11 | 0.26 | 171.40 | 5.14 | 0.26 |
| 10.00 | 131.37 | 4.87 | 0.26 | 126.51 | 4.84 | 0.29 |
| 12.00 | 106.96 | 4.67 | 0.30 | 104.97 | 4.65 | 0.31 |
| 24.00 | 34.00 | 3.52 | 0.40 | 34.03 | 3.52 | 0.44 |
| 48.00 | 2.54 | 0.93 | 0.56 | 1.64 | 0.49 | 0.90 |
| 72.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.0 |
Plasma concentration (mean±SD) per h (0–72 h) in 23 subjects. SD: Standard deviation
In accordance with the FDA Empagliflozin Guidelines (2015),[7] we performed a single-dose, two-treatment, two-period crossover study in vivo under the fasted condition utilizing a high dose of empagliflozin 25 mg. Before this, we conducted an acceptable dissolution test in vitro to confirm the theoretical consistency of the anticipated results between T and R formulation.[17] To avoid hypoglycemic episodes, the empagliflozin was administered with 240 mL of a 20% glucose solution in water, followed by 60 mL of the glucose solution administered every 15 min for up to 4 h after of R or T oral simple doses. Standard meal was provided at 4, 8, 12, and 24 h after doses. No AEs were reported.
Limitations
Per study protocol, both male and female subjects were to be enrolled in the study, but only male subjects were included due to limited recruitment criteria and were not possible to include female volunteers.[18] Empagliflozin is not recommended for women trying to conceive because is considered a pregnancy safety category C.[1,5,6]
An acceptable fasting BE study alone does not meet the requirements for approval of empagliflozin tablets under the guidelines of some regulatory agencies. According to US FDA guidelines, 2-period, 2-sequence, and crossover BE studies under fasting and fed conditions are required to determine that the 90% CI of T/R ratios are within the range of 80–125% for Cmax and AUC.[7]
CONCLUSION
Our study demonstrated that the test formulation Izaban® 25 mg tablets of Laboratorios Leti S.A.V. is bioequivalent and well tolerated as the reference formulation Jardiance® 25 mg tablets of Boehringer Ingelheim Pharmaceuticals, Inc., in healthy adult male subjects under fasting conditions. According to the US FDA Empagliflozin Guidelines, the PK profiles were within the BE range (80.00–125.00%). With more availability of generic versions of effective pharmacotherapeutic agents[18] such as empagliflozin tablets on the global market, the cost of treatment of patients with diabetes will be low and affordable to patients and the governments.
Acknowledgments
This study was conducted at the third party ICBio Clinical Research Pvt. Ltd, located in Vidyaranyapura, Bangalore, India.
Author’s contributions
EP, AI, AT, XS, and JGC performed the statistical analysis, interpretation, writing, revision, of the manuscript.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
All authors are employees of Industrias Biocontrolled C.A., (Leti Group Company), and may hold share and/or stock options in the company. The authors have no other potential conflicts of interest relevant to this study.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
This study was funded by Laboratorios Leti S.A.V.
References
- Empagliflozin: A review in type 2 diabetes. Drugs. 2018;78:1037-1048. doi:10.1007/s40265-018-0937-z
- [CrossRef] [PubMed] [Google Scholar]
- Management of hyperglycemia in type 2 diabetes, 2015: A patient-centered approach: Update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140-149. doi:10.2337/dc14-2441
- [CrossRef] [PubMed] [Google Scholar]
- Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: From pharmacology to pre-clinical and clinical therapeutics. Theranostics. 2021;11:4502-4515. doi:10.7150/thno.54498
- [CrossRef] [PubMed] [Google Scholar]
- SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol. 2019;18:15. doi:10.1186/s12933-019-0816-2
- [CrossRef] [PubMed] [Google Scholar]
- Jardiance® (empagliflozin) tablets, for oral use. 2016. US prescribing information. Available from: https://docs.boehringeringelheim.com/prescribing%20information/pis/jardiance/jardiance.pdf [Last accessed on 2023 Jun 08]
- [Google Scholar]
- Jardiance 10 and 25 mg film-coated tablets: Summary of product characteristics. 2014. Available from: https://health.ec.europa.eu/system/files/2016-11/smpc_guideline_rev2_en_0.pdf [Last accessed on 2023 Jul 12]
- [Google Scholar]
- FDA product-specific guidances for generic drug development web page. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/psg/empagliflozin_oral%20tablet_204629_rc06-15.pdf [Last accessed on 2023 Jun 08]
- [Google Scholar]
- ICMR guidelines. 2017. Available from: https://main.icmr.nic.in/sites/default/files/guidelines/icmr_ethical_guidelines_2017.pdf [Last accessed on 2023 Jul 12]
- [Google Scholar]
- New drugs and clinical trials rules 2019: Changes in responsibilities of the ethics committee. Perspect Clin Res. 2020;11:37-43. doi:10.4103/picr.PICR_208_19
- [CrossRef] [PubMed] [Google Scholar]
- World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. Bull World Health Organ. 2001;79:373-374. doi:10.1001/jama.2013.281053
- [CrossRef] [PubMed] [Google Scholar]
- E6: Guidance on good clinical practice. 2002. PMP/ICH/135/95 European Medicines Agency. Available from: https://www.imim.cat/media/upload/arxius/emea.pdf [Last accessed on 2023 Jul 08]
- [Google Scholar]
- Pharmacokinetic evaluation of empagliflozin in healthy Egyptian volunteers using LC-MS/MS and comparison with other ethnic populations. Sci Rep. 2017;7:2583. doi:10.1038/s41598-017-02895-7
- [CrossRef] [PubMed] [Google Scholar]
- Bioavailability and bioequivalence studies In: Pharmaceutical Formulation Design-recent Practices. United Kingdom: IntechOpen; 2020. doi:10.5772/intechopen.85145
- [CrossRef] [Google Scholar]
- Pharmacokinetics and bioequivalence of two empagliflozin, with evaluation in healthy Jordanian subjects under fasting and fed conditions. Pharmaceuticals (Basel). 2022;15:193. doi:10.3390/ph15020193
- [CrossRef] [PubMed] [Google Scholar]
- Comparison of the pharmacokinetics, safety, and tolerability of two empagliflozin formulations in healthy Korean subjects. Drug Des Dev Ther. 2023;17:2137-2145. doi:10.2147/DDDT.S409368
- [CrossRef] [PubMed] [Google Scholar]
- Triple fixed-dose combination empagliflozin, linagliptin, and metformin for patients with type 2 diabetes. Postgrad Med. 2020;132:337-345. doi:10.1080/00325481.2020.1750228
- [CrossRef] [PubMed] [Google Scholar]
- Guía para la Industria: Pruebas de disolucion de formas de dosificacion oral solidas de liberacion inmediata. 2018. FDA. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guía-para-la-industriapruebas-de-disolución-de-formas-de-dosificación-oral-solidas-de-liberación [Last accessed on 2023 Jul 20]
- [Google Scholar]
- Bioequivalence study of two formulations of rivaroxaban in healthy adult subjects under fasting conditions. Am J Pharmacother Pharm Sci. 2023;2:8. doi:10.25259/AJPPS_2023_008
- [CrossRef] [Google Scholar]




